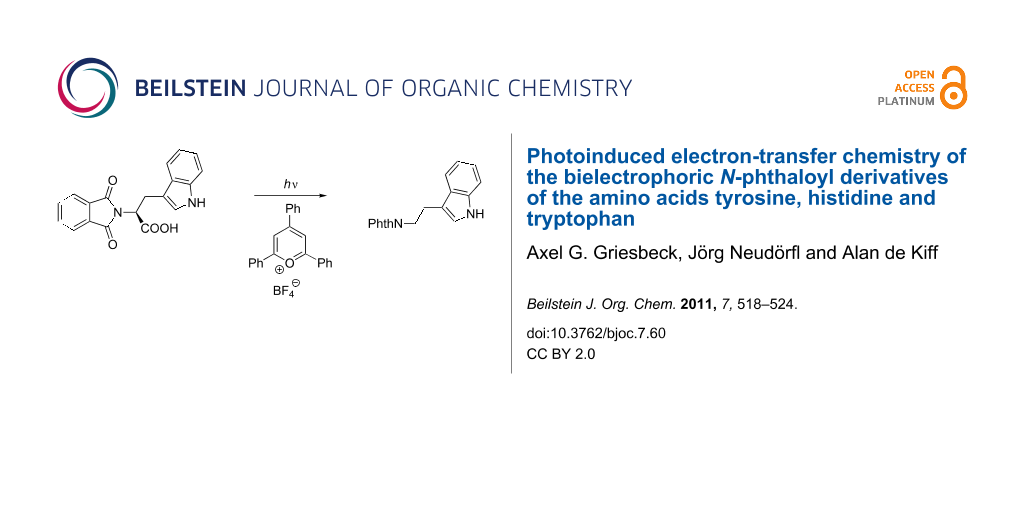
Abstract
The photochemistry of phthalimide derivatives of the electron-rich amino acids tyrosine, histidine and tryptophan 8–10 was studied with respect to photoinduced electron-transfer (PET) induced decarboxylation and Norrish II bond cleavage. Whereas exclusive photodecarboxylation of the tyrosine substrate 8 was observed, the histidine compound 9 resulted in a mixture of histamine and preferential Norrish cleavage. The tryptophan derivative 10 is photochemically inert and shows preferential decarboxylation only when induced by intermolecular PET.

Graphical Abstract
Introduction
Phthalimides are versatile electron acceptors in photoinduced electron-transfer (PET) reactions. N-Alkylated phthalimides typically absorb in the 295 nm region with extinction coefficients around 103. The quantum yields for intersystem crossing ФISC significantly change with the substitution on the imide nitrogen, e.g., ФISC = 0.5 for N-isobutylphthalimide and ФISC < 0.01 for N-arylphthalimides [1]. If necessary, the population of the triplet state is also possible by sensitization, e.g., with triplet sensitizers such as acetone or benzophenone. With a triplet energy ET of 293–300 kJ mol−1 and a ground-state reduction potential E0 of −1.85 V vs Fc/Fc+, electronically excited phthalimides are potent electron acceptors [2]. The rich photochemistry of this chromophore has recently been reviewed [3,4]. Intramolecular hydrogen abstraction is an archetype process for electronically excited carbonyl groups (Norrish type II reaction). The 1,4-biradicals formed by γ-CH transfer can undergo several subsequent reactions, among which are secondary H transfer, cyclization, or fragmentation. The excited imido group is at the same time an efficient electron acceptor and can be reduced by numerous donor groups.
As a model compound for neutral aliphatic amino acids, the photophysical and photochemical properties of N-phthaloylvaline methyl ester (1) have been studied by nanosecond laser flash photolysis (λexc = 248 or 308 nm) [5]. The quantum yield of fluorescence is low (ФF = 10−2), whereas that of phosphorescence at −196 °C is large (0.5). The triplet properties of 1 at room temperature and in ethanol at low temperatures are known: Triplet acetone, acetophenone and xanthone in acetonitrile are quenched by 1 via energy transfer; the rate constant is almost diffusion-controlled and somewhat smaller for benzophenone. The sole product from the photolysis of 1 is the double hydrogen transfer product 2. On the other hand, phthaloyl derivatives of C-unprotected α-amino acids (e.g., derivatives of Gly, Ala, Val, Ile, Phe) undergo efficient photodecarboxylation to yield the corresponding amines, β-amino acids are converted to benzazepines, and γ-amino acids to benzopyrrolizidines (Scheme 1). For example, the glutamic acid derivative 3 resulted in the formation of a diastereoisomeric mixture of benzopyrrolizidinones 4 [6]. For a specific system, N-phthaloyl methionine (5), we have detected bielectrophoric behavior in that the electron transfer from the thioether group competes efficiently with decarboxylation when the reaction is triplet-sensitized [7]. Photodecarboxylation is followed by PET cyclization to give 6, whereas the lactone 7 originates from PET-induced sulfur oxidation and radical ion trapping without subsequent decarboxylation. Similar effects were observed for the cysteine and S-methyl cysteine derivatives [8].
![[1860-5397-7-60-i1]](/bjoc/content/inline/1860-5397-7-60-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Three phthalimide/amino acid model reactions: Norrish II process of 1, PET decarboxylation of 3, PET competition of 5.
Scheme 1: Three phthalimide/amino acid model reactions: Norrish II process of 1, PET decarboxylation of 3, PE...
Other proteinogenic amino acids that, in principle, should also be able to show bielectrophoric behavior with aromatic side chains similar to phenylalanine are tyrosine, histidine and tryptophan. The photochemistry of the phthalimide derivatives of these three amino acids are described in this publication.
Results
Synthesis of phthalimide substrates
The C-unprotected N-phthaloyl amino acids 8–10 were available from tyrosine, histidine, and tryptophan (Figure 1). The phthalimide derivatives were prepared either by thermal reaction of phthalic anhydride with the corresponding amino acids or by the Nefkens procedure [9]. The latter procedure yields enantiomerically pure products (by optical rotation), whereas the thermal method leads to partial epimerization.
![[1860-5397-7-60-1]](/bjoc/content/figures/1860-5397-7-60-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phthalimides from tyrosine 8, histidine 9 and tryptophan 10.
Figure 1: Phthalimides from tyrosine 8, histidine 9 and tryptophan 10.
Photochemistry of the tyrosine and histidine derivatives 8 and 9
The colorless phthalimides from tyrosine and histidine 8 and 9, respectively, were photochemically active and gave decarboxylation and cleavage products. In contrast to photolysis in pure acetone [10], irradiation of the tyrosine substrate 8 in basic water/acetone resulted solely in the decarboxylation product 11. Under identical conditions, the histidine derivative 9 resulted in a 1:4 mixture of phthaloyl histamine 12 and phthalimide (13) (Scheme 2). In both cases, the photolyses were clean processes and the products were isolated in high yields after complete conversion. The tyramine derivative 11 was isolated by crystallization in near quantitative yield.
![[1860-5397-7-60-i2]](/bjoc/content/inline/1860-5397-7-60-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: PET decarboxylation/photocleavage of 8 and 9.
Scheme 2: PET decarboxylation/photocleavage of 8 and 9.
Photochemistry of the tryptophan derivative 10
In contrast to all other – colorless – phthalimides of the proteinogenic amino acid series, the tryptophan derivative 10 is bright yellow. This compound, which we crystallized and whose solid-state structure was determined by X-ray diffraction analysis (Figure 2) [11], has also been described in literature as a yellow compound [12,13]. The color is not the consequence of any impurity and did not vanish after several recrystallizations and, more importantly, was also observed for the methyl ester and the decarboxylation product, the N-phthaloyl tryptamine (14). Compound 10 represents a donor–ethylene bridge–acceptor situation and intramolecular electron transition (ET) is feasible. The UV spectra of the three substrates 8–10 (Figure 3) showed in fact two major absorption peaks for 8 and 10 at 280 and 300 nm, whereas the histidine derivative 9 only exhibits the typical phthalimide absorption at λmax = 282 nm. The red-shifted absorption of 10 has a tailing that explains the color of this compound and derivatives as CT state absorption and is additionally red-shifted by 10 nm on changing the solvent from ether to acetonitrile or methanol, respectively. It is remarkable that under solid-state conditions the molecular conformation of 10 in the crystal lattice is synclinal with respect to the indole and the phthalimide groups as well as for the indole and the carboxy groups, indicating a ground-state electronic interaction between the donor indole and the acceptor phthalimide (Figure 2). Such interactions also in solution phase would facilitate decay processes from the excited singlet or triplet state. This concept has also been described by Gawronski et al. from exciton CD Cotton effects that have been measured for a series of bichromophoric systems based on phthalimide–linker–donor triades [12]. Concerning the photochemistry of 10, a remarkable difference to all other phthalimide derivatives occurred: Neither direct excitation nor triplet (acetone, benzophenone) sensitized conditions led to substrate conversion even after prolonged irradiation. Only the use of the PET catalyst 2,4,6-triphenylpyrylium tetrafluoroborate enabled quantitative conversion to give a 4:1 mixture of the decarboxylation product, N-phthaloyl typtamine (14) and the cleavage product 13 (Scheme 3).
![[1860-5397-7-60-2]](/bjoc/content/figures/1860-5397-7-60-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of the tryptophan derivative 10 in the crystal.
Figure 2: Structure of the tryptophan derivative 10 in the crystal.
![[1860-5397-7-60-3]](/bjoc/content/figures/1860-5397-7-60-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis absorption spectra of compounds 8–10 (c = 2 × 10−4 in CH3OH).
Figure 3: UV–vis absorption spectra of compounds 8–10 (c = 2 × 10−4 in CH3OH).
![[1860-5397-7-60-i3]](/bjoc/content/inline/1860-5397-7-60-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Direct, triplet-sensitized and ET-sensitized photochemistry of 10.
Scheme 3: Direct, triplet-sensitized and ET-sensitized photochemistry of 10.
Additionally, the fluorescence spectra (Figure 4) indicated a charge-transfer excited-state formation from the tyrosine and tryptophan derivatives, 8 and 10, respectively, by showing dual emission at 310 and 370 nm.
![[1860-5397-7-60-4]](/bjoc/content/figures/1860-5397-7-60-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorescence spectra of compounds 8–10 (c = 6 × 10−5 in CH3OH).
Figure 4: Fluorescence spectra of compounds 8–10 (c = 6 × 10−5 in CH3OH).
Discussion
Two reaction modes were observed with the phthalimides 8–10: (a) Norrish type II cleavage of the central N–C bond to give the N-unsubstituted phthalimide 13, and (b) photochemical decarboxylation. Analysis of the energetics of the PET between the phthalimide acceptor (see Introduction) and the arene donor side chains of the corresponding amino acid leads to the conclusion that PET is exergonic with ΔG = −0.1 to −0.3 V for all cases: The redox potentials of the three amino acids tryptophan, histidine and tyrosine are reported to be 1.02 V [14], 1.17 V [15] and 0.93 V [16].
Due to this narrow redox potential window, similar electron-transfer properties can be expected. Among the amino acids investigated herein, tryptophan is described in biological electron-transfer processes as the most active hole transport molecule [17]. Thus, it can be concluded that the excited charge-transfer state from 10, as can be interpreted from its fluorescence spectrum, is rapidly deactivated radiatively as well as non-radiatively by back electron transfer (BET). In contrast to 10, electron transfer from the electronically excited tyrosine compound 8 is followed by oxidation of the carboxyl anion and subsequent decarboxylation to give N-phthaloyl tyramine (11). If BET from the radical cation state of 10 can be retarded (i.e., produced by an intermolecular PET), the corresponding decarboxylation proceeds to give N-phthaloyl tryptamine (14) (Scheme 3). The histidine derivative 9 is expected to undergo PET with oxidation of the aryl group with the lowest efficiency and thus a Norrish II process (γ-hydrogen transfer with subsequent central bond cleavage) prevails here (Scheme 4). This process, which was also detected as the minor pathway for the histidine case can, of course, also be described as an electron-transfer induced process followed by α-deprotonation.
![[1860-5397-7-60-i4]](/bjoc/content/inline/1860-5397-7-60-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
The photochemistry of three bielectrophoric phthaloyl derivatives of tyrosine (8), histidine (9) and tryptophan (10) follows two distinct pathways, fragmentation and decarboxylation. Both routes can be described as initiated by electron transfer. Intramolecular transfer in the tyrosine substrate 8 leads to efficient decarboxylation whereas for the tryptophan substrate 10 this process can only be initiated by intermolecular electron transfer suggesting efficient back electron transfer in the photoexcited substrate.
Experimental
Synthesis of N-phthaloyl tyrosine (8) [18]. A well ground mixture of 2.00 g (13 mmol) of L-tyrosine and 2.45 g (13 mmol) of phthalic anhydride were added to a 10 mL flask with a magnetic stirring bar. The flask was heated in an oil bath, stirred at 150–160 °C for 20 min and then the mixture was allowed to cool to room temperature. Recrystallization of the crude solid material from ethanol gave 3.67 g of N-phthaloyl tyrosine (8) (88%) as colorless needles; mp 161–163 °C (lit. 162–164 °C); [α]D20 −183.8 (c 1.0, EtOH) (lit. −182.4); 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 3.22 (dd, 2H, J1 = 13.6 Hz, J2 = 11.8 Hz), 5.01 (dd, 1H, J1 = 11.9 Hz, J2 = 5.0 Hz), 6.53 (d, 1H, J = 8.5 Hz), 6.91 (d, J = 8.5 Hz), 7.84 (s, 4H), 9.14 (s, COOH). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 33.1 (t, 1C), 53.2 (s, 1C), 115.1 (d, 2C), 123.4 (d, 2C), 127.2 (s, 1C), 129.7 (d, 2C), 130.8 (s, 2C), 134.9 (d, 2C), 155.8 (s, 1C), 167.1 (s, 2C), 170.2 (s, 1C).
Synthesis of N-phthaloyl histidine (9) [19]. L-histidine 3.0 g (14 mmol) and 1.52 g (14 mmol) of Na2CO3 were dissolved in 100 mL water and 3.14 g (14 mmol) of N-carbethoxy phthalimide was added to the solution. The mixture was allowed to stir at room temperature for 2 h. After acidification with 2 M HCl, the solvent was removed under reduced pressure. The colorless residue was heated under reflux in 10 mL MeOH for 20 min. After cooling, the residue was removed by filtration and washed with 100 mL MeOH to give 3.40 g of N-phthaloyl histidine (9) (83%) as a colorless solid; mp >260 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 3.37 (d, 2H, J = 6.6 Hz, H 7), 4.91 (dd, 1H, J1 = 9.5 Hz, J2 = 6.5 Hz), 6.81 (s, 1H), 7.73 (s, 1H), 7.84 (s, 4H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 33.1 (t, 1C), 53.2 (s, 1C), 115.1 (d, 2C), 123.4 (d, 2C), 127.2 (s, 1C), 129.7 (d, 2C), 130.8 (s, 2C), 134.9 (d, 2C), 155.8 (s, 1C), 167.1 (s, 2C), 170.2 (s, 1C).
Synthesis of N-phthaloyl tryptophan (10) [18]. A mixture of 2 g (9.8 mmol) of L-tryptophan and 1.04 g (9.8 mmol) of Na2CO3 was dissolved in 100 mL of water. To this solution, 2.15 g (9.8 mmol) of N-carbethoxy phthalimide were added. The mixture was stirred at room temperature for 1 h. After filtration the solution was acidified with 2 M HCl and the precipitate collected. Recrystallization from aqueous acetone gave 3.12 g of N-phthaloyl tryptophan (10) (95%) as yellow needles; mp 170 °C; [α]D20 −247.5 (c 1.0, EtOH) (lit: −249.6); 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 3.55–3.62 (m, 2H), 5.13 (dd, 1H, J1 = 10 Hz, J2 = 6.7 Hz), 6.89 (t, 1H, J = 7.3 Hz), 7.00 (t, 1H, J = 8 Hz), 7.49 (d, 1H, J = 8.1 Hz), 7.26 (d, 1H, J = 8.2 Hz), 7.80 (s, 4H), 10.74 (s, COOH). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 24.1 (t, 1C), 52.7 (d, 1C), 109.8 (s, 1C), 111.5 (d, 1C), 117.9 (d, 1C), 118.5 (d, 1C), 121.0 (d, 1C), 123.4 (d, 2C), 126.9 (s, 1C), 130.9 (s, 2C), 134.9 (d, 2C), 136.1 (s, 1C), 167.2 (s, 2C), 170.4 (s, 1C).
Photolysis of N-phthaloyl tyrosine (8). A water-cooled solution (c = 2.1 × 10−3 mol/l) of 8 and 0.5 equiv K2CO3 in 100 mL of an acetone/water mixture (1:1) was irradiated at 300 nm (lamps: 8 × 3000 Å, 800 W, 300 ± 10 nm) for 12 h under a nitrogen atmosphere. The product was removed by filtration and washed with water to give 396 mg of N-phthaloyl tyramine (11) (95%) as a colorless solid; mp 171–174 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 2.77 (t, 2H, J = 7.5 Hz), 3.73 (t, 2H, J = 7.5 Hz), 6.59 (d, 2H, J = 8.5 Hz), 6.92 (d, 2H, J = 8.5 Hz), 7.82 (s, 2H), 7.83 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 32.9 (t, 1C), 40.3 (t, 1C), 115.5 (d, 2C), 123.0 (d, 2C), 127.0 (s, 1C), 129.4 (d, 2C, C8/C8’), 131.5 (s, 2C), 134.4 (d, 2C), 157.2 (s, 1C), 167.7 (s, 2C).
Photolysis of N-phthaloyl histidine (9). A water-cooled solution (c = 2.1 × 10−3 mol/l) of 9 and 0.5 equiv K2CO3 in 100 mL of an acetone/water mixture (1:1) was irradiated at 300 nm (lamps: 8 × 3000 Å, 800 W, 300 ± 10 nm) under a nitrogen atmosphere. After 24 h the solution was acidified with 2 M HCl and the solvent was removed under reduced pressure. The product composition [N-phthaloyl histamine (12)/phthalimide (13)] was determined by 1H NMR spectroscopy.
12: 20% (by 1H NMR); mp 173–175 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 2.91 (t, 2H, J = 6.6 Hz), 3.88 (t, 2H, J = 6.6 Hz), 6.79 (s, 1H), 7.70 (s, 1H), 7.80 (s, 4H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 24.0 (t, 1C), 37.9 (d, 1C, C5), 116.1 (d, 1C), 123.1 (d, 2C), 131.1 (s, 2C), 132.9 (d, 1C), 134.5 (d, 2C), 138.4 (s, 1C), 167.9 (s, 2C, C4/C4’).
13: 80% (by 1H NMR); 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 7.75 (s, 4H). 13C NMR (75 MHz, DMSO-d6): δ (ppm) = 123.0 (d, 2C), 132.1 (s, 2C), 134.7 (d, 2C), 167.2 (s, 2C).
Photolysis of N-phthaloyl tryptophan (10). A 100 mL solution (c = 0.01 mol/l) of 10 and 5.4 × 10−3 mmol 2,4,6-triphenylpyrylium tetrafluoroborate in acetonitrile was irradiated at 350 nm (RPR-3500 Å lamp: 6 × 3500 Å, 400 W, 350 ± 25 nm) for 4 d under a nitrogen atmosphere. After removal of the solvent under reduced pressure, the ratio of products was determined by 1H NMR spectroscopy. Silica gel flash chromatography (cyclohexane/ethylacetate) of the residue afforded N-phthaloyl tryptamine (14) as a yellow solid and phthalimide (13) as a colorless solid.
14: 80% (by 1H NMR); mp 173–175 °C; 1H NMR (300 MHz, acetone-d6): δ (ppm) = 3.13 (t, 2H, J = 7.7 Hz), 3.95 (t, 2H, J = 7.7 Hz), 7.01 (t, 1H, J = 7.4 Hz), 7.09 (t, 1H, J = 7.8 Hz), 7.20 (s, 1H), 7.37 (d, 1H, J = 7.9 Hz), 7.67 (d, 1H, J = 87.7 Hz), 7.81 (s, 4H). 13C NMR (75 MHz, acetone-d6): δ (ppm) = 24.2 (t, 1C), 38.4 (t, 1C), 111.4 (d, 1C), 118.3 (d, 1C), 118.7 (d, 1C), 121.3 (d, 1C), 122.9 (d, 2C), 127.6 (s, 1C), 132.3 (s, 2C), 134.1 (d, 2C), 135.9 (s, 1C), 167.9 (s, 2C).
References
-
Warzecha, K.-D.; Görner, H.; Griesbeck, A. G. J. Phys. Chem. A 2006, 110, 3356–3363. doi:10.1021/jp055878x
Return to citation in text: [1] -
Wintgens, V.; Valat, P.; Kossanyi, J.; Biczok, L.; Demeter, A.; Bérces, T. J. Chem. Soc., Faraday Trans. 1994, 90, 411–421. doi:10.1039/ft9949000411
Return to citation in text: [1] -
Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2001, 34, 523–532. doi:10.1021/ar010004o
Return to citation in text: [1] -
Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w
Return to citation in text: [1] -
Griesbeck, A. G.; Görner, H. J. Photochem. Photobiol., A: Chem. 1999, 129, 111–119. doi:10.1016/S1010-6030(99)00180-X
Return to citation in text: [1] -
Griesbeck, A. G.; Nerowski, F.; Lex, J. J. Org. Chem. 1999, 64, 5213–5217. doi:10.1021/jo990390b
Return to citation in text: [1] -
Griesbeck, A. G.; Mauder, H.; Müller, I.; Peters, K.; Peters, E.-M.; von Schnering, H. G. Tetrahedron Lett. 1993, 34, 453–456. doi:10.1016/0040-4039(93)85100-B
Return to citation in text: [1] -
Griesbeck, A. G.; Hirt, J.; Kramer, W.; Dallakian, P. Tetrahedron 1998, 54, 3169–3180. doi:10.1016/S0040-4020(98)00063-5
Return to citation in text: [1] -
Nefkens, G. H. L.; Tesser, G. I.; Nivard, R. J. F. Recl. Trav. Chim. Pays-Bas 1960, 79, 688–698. doi:10.1002/recl.19600790705
Return to citation in text: [1] -
Griesbeck, A. G.; Henz, A.; Hirt, J.; Ptatschek, V.; Engel, T.; Löffler, D.; Schneider, F. W. Tetrahedron 1994, 50, 701–714. doi:10.1016/S0040-4020(01)80787-0
Return to citation in text: [1] -
The crystallographic data for the tryptophan derivative 10 has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-784387.
Return to citation in text: [1] -
Gawronski, J.; Kazmierczak, F.; Gawronska, K.; Skowronek, P.; Waluk, J.; Marczyk, J. Tetrahedron 1996, 52, 13201–13214. doi:10.1016/0040-4020(96)00795-8
Return to citation in text: [1] [2] -
Casimir, J. R.; Guichard, G.; Briand, J.-P. J. Org. Chem. 2002, 67, 3764–3768. doi:10.1021/jo016347h
Return to citation in text: [1] -
Harriman, A. J. Phys. Chem. 1987, 91, 6102–6104. doi:10.1021/j100308a011
Return to citation in text: [1] -
Navaratnam, S.; Parsons, B. J. J. Chem. Soc., Faraday Trans. 1998, 94, 2577–2581. doi:10.1039/a803477j
Return to citation in text: [1] -
Cordes, M.; Köttgen, A.; Jasper, C.; Jacques, O.; Boudebous, H.; Giese, B. Angew. Chem., Int. Ed. 2008, 47, 3461–3463. doi:10.1002/anie.200705588
Return to citation in text: [1] -
Giese, B.; Wang, M.; Stoltz, M.; Müller, P.; Graber, M. J. Org. Chem. 2009, 74, 3621–3625. doi:10.1021/jo900375f
Return to citation in text: [1] -
Zeng, Q.; Liu, Z.; Li, B.; Wang, F. Amino Acids 2004, 27, 183–186. doi:10.1007/s00726-004-0109-1
Return to citation in text: [1] [2] -
Auterhoff, H.; Hansen, J.-G. Pharmazie 1970, 25, 336–340.
Return to citation in text: [1]
| 18. | Zeng, Q.; Liu, Z.; Li, B.; Wang, F. Amino Acids 2004, 27, 183–186. doi:10.1007/s00726-004-0109-1 |
| 18. | Zeng, Q.; Liu, Z.; Li, B.; Wang, F. Amino Acids 2004, 27, 183–186. doi:10.1007/s00726-004-0109-1 |
| 1. | Warzecha, K.-D.; Görner, H.; Griesbeck, A. G. J. Phys. Chem. A 2006, 110, 3356–3363. doi:10.1021/jp055878x |
| 6. | Griesbeck, A. G.; Nerowski, F.; Lex, J. J. Org. Chem. 1999, 64, 5213–5217. doi:10.1021/jo990390b |
| 16. | Cordes, M.; Köttgen, A.; Jasper, C.; Jacques, O.; Boudebous, H.; Giese, B. Angew. Chem., Int. Ed. 2008, 47, 3461–3463. doi:10.1002/anie.200705588 |
| 5. | Griesbeck, A. G.; Görner, H. J. Photochem. Photobiol., A: Chem. 1999, 129, 111–119. doi:10.1016/S1010-6030(99)00180-X |
| 17. | Giese, B.; Wang, M.; Stoltz, M.; Müller, P.; Graber, M. J. Org. Chem. 2009, 74, 3621–3625. doi:10.1021/jo900375f |
| 3. | Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2001, 34, 523–532. doi:10.1021/ar010004o |
| 4. | Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w |
| 2. | Wintgens, V.; Valat, P.; Kossanyi, J.; Biczok, L.; Demeter, A.; Bérces, T. J. Chem. Soc., Faraday Trans. 1994, 90, 411–421. doi:10.1039/ft9949000411 |
| 15. | Navaratnam, S.; Parsons, B. J. J. Chem. Soc., Faraday Trans. 1998, 94, 2577–2581. doi:10.1039/a803477j |
| 10. | Griesbeck, A. G.; Henz, A.; Hirt, J.; Ptatschek, V.; Engel, T.; Löffler, D.; Schneider, F. W. Tetrahedron 1994, 50, 701–714. doi:10.1016/S0040-4020(01)80787-0 |
| 12. | Gawronski, J.; Kazmierczak, F.; Gawronska, K.; Skowronek, P.; Waluk, J.; Marczyk, J. Tetrahedron 1996, 52, 13201–13214. doi:10.1016/0040-4020(96)00795-8 |
| 13. | Casimir, J. R.; Guichard, G.; Briand, J.-P. J. Org. Chem. 2002, 67, 3764–3768. doi:10.1021/jo016347h |
| 9. | Nefkens, G. H. L.; Tesser, G. I.; Nivard, R. J. F. Recl. Trav. Chim. Pays-Bas 1960, 79, 688–698. doi:10.1002/recl.19600790705 |
| 12. | Gawronski, J.; Kazmierczak, F.; Gawronska, K.; Skowronek, P.; Waluk, J.; Marczyk, J. Tetrahedron 1996, 52, 13201–13214. doi:10.1016/0040-4020(96)00795-8 |
| 8. | Griesbeck, A. G.; Hirt, J.; Kramer, W.; Dallakian, P. Tetrahedron 1998, 54, 3169–3180. doi:10.1016/S0040-4020(98)00063-5 |
| 7. | Griesbeck, A. G.; Mauder, H.; Müller, I.; Peters, K.; Peters, E.-M.; von Schnering, H. G. Tetrahedron Lett. 1993, 34, 453–456. doi:10.1016/0040-4039(93)85100-B |
| 11. | The crystallographic data for the tryptophan derivative 10 has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-784387. |
© 2011 Griesbeck et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)