Abstract
We describe the unprecedented formation of six ansamitocin derivatives that are deoxygenated at C-7 of the ansamitocin core, obtained during fermentation experiments by employing a variety of Actinosynnema pretiosum mutants and mutasynthetic approaches. We suggest that the formation of these derivatives is based on elimination at C-7/C-8 followed by reduction(s) of the intermediate enone. In bioactivity tests, only ansamitocin derivatives bearing an ester side chain at C-3 showed strong antiproliferative activity.

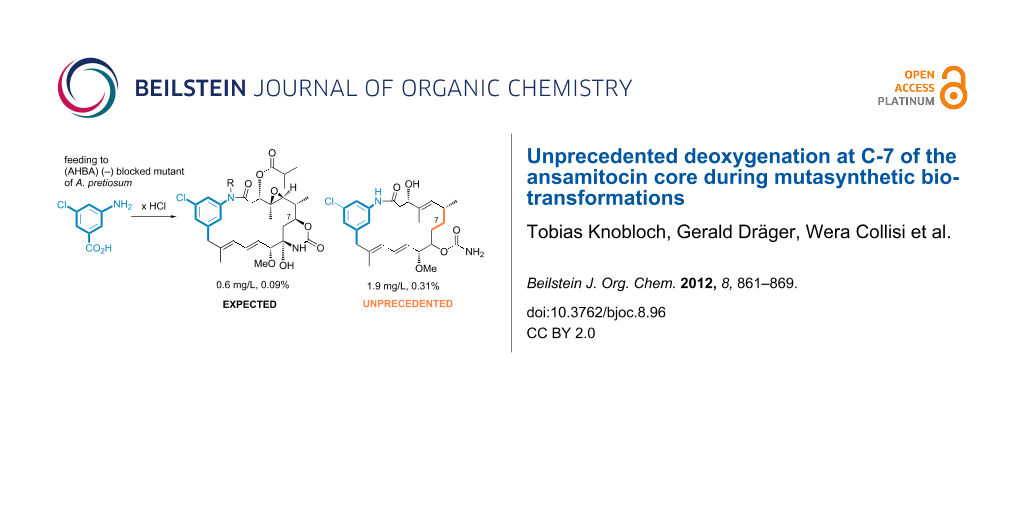
Graphical Abstract
Introduction
Natural products still play an important role as lead structures for the treatment of infectious diseases and cancer. However, natural products have lost some of their attraction for the development of pharmaceuticals because of their structural complexity and the difficulties associated with accessing analogues for structure–activity relationship studies. Total synthesis approaches are still a tour de force and are hardly employed, while commonly semisynthesis as well as biotechnological approaches are widely pursued in industrial research [1-3]. Investigations into the biosynthesis of natural products have not only allowed us to understand the synthetic principles that nature pursues, but have also provided tools, mainly based on genetic engineering, which can be exploited in natural product synthesis [4].
Producer strains genetically blocked in the biosynthesis of important and complex natural products can serve as such new tools. The synthetic concept based on these blocked mutants is termed “mutational biosynthesis”, or in short mutasynthesis, and it relies on the cellular uptake of modified biosynthetic intermediates, sometimes termed mutasynthons, and their incorporation into complex secondary metabolites [5-7].
When making use of mutants that are blocked in early stages of a given biosynthesis pathway, the concept of mutasynthesis may be compared to a (partial) natural product total synthesis. When further modification of an advanced biosynthetic intermediate with an established core structure towards bioactive natural products and analogues is conducted, mutasynthesis may be regarded as the “endgame” of a total synthesis [4].
The ansamitocins (maytansinoids) 3–5 are ideally suited for mutasynthetic modifications and the creation of new analogues because they are highly potent antitumor active compounds that inhibit the growth of different leukemia cell lines as well as human solid tumors at very low concentrations (10−3 to 10−7 µg/mL) [8]. In contrast to colchicine, maytansinoids such as ansamitocins bind to β-tubulin monomers at a site overlapping the vinca alkaloid binding site [9].
Recently, we disclosed several mutasynthetic studies aimed at the production of derivatives of ansamitocins 3–5 [10-12] as well as of geldanamycin (6), utilizing mutant strains of Actinosynnema pretiosum, the ansamitocin producer [13-17], and Streptomyces hygroscopicus, the geldanamycin producer [18,19]. These engineered strains are unable to biosynthesize 3-amino-5-hydroxybenzoic acid (1) [20], the common starter unit for both polyketide synthases (PKS) (Scheme 1). These assembly-line-type multienzymes are responsible for setting up the complete carbon backbone of both ansamycin antibiotics [21-24].
![[1860-5397-8-96-i1]](/bjoc/content/inline/1860-5397-8-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Summary of ansamitocin biosynthesis and structure of the related ansamycin antibiotic geldanamycin (6).
Scheme 1: Summary of ansamitocin biosynthesis and structure of the related ansamycin antibiotic geldanamycin (...
More precisely, the biosynthesis of ansamitocins relies on a type I modular polyketide synthase (PKS), with 3-amino-5-hydroxybenzoic acid (1, AHBA) [20] as the starter unit followed by chain extension by one “glycolate”, three propionate and three acetate units. The last PKS module holds seco-proansamitocin, which is released and cyclized, presumably by an ansamycin amide synthase (Asm9) [21-24], to yield the 19-membered macrocyclic lactam proansamitocin (2). Proansamitocin (2) is transformed into bioactive compounds 3–5 by a set of post-PKS tailoring steps, following a predetermined, only partly flexible logic (Scheme 1) [16].
Complementing our studies with mutant strain A. pretiosum HGF073, blocked in the biosynthesis of the PKS starter unit AHBA 1 [13-16], we recently reported the use of a mutant of A. pretiosum blocked in Asm12 (chlorination) and Asm21 (carbamoylation) and therefore producing proansamitocin (2) in good yield (up to 106 mg/L of fermentation broth) [17]. Additionally, we isolated small amounts of O-methyl proansamitocin 7 (2.3 mg/L), 10-epi-proansamitocin 8 (3.5 mg/L) and two diastereomeric byproducts 9a and 9b (7.6 mg/L; 1:1 ratio) from the fermentation broth of A. pretiosum Δasm12/21 (Figure 1). We also showed that none of these proansamitocin derivatives exhibit antiproliferative activity.
![[1860-5397-8-96-1]](/bjoc/content/figures/1860-5397-8-96-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Fermentation products, proansamitocin (2) and derivatives 7–9, of the Asm12 and Asm21-blocked (chlorination, carbamoylation) mutant strain A. pretiosum Δasm12/21 (yields given as isolated product per volume of fermentation broth) [17].
Figure 1: Fermentation products, proansamitocin (2) and derivatives 7–9, of the Asm12 and Asm21-blocked (chlo...
Herein, we describe the unprecedented formation of ansamitocin derivatives that are deoxygenated at C-7 of the ansamitocin core, obtained by us during fermentation experiments using a variety of A. pretiosum mutants and mutasynthetic approaches.
Results and Discussion
In the course of our well-established mutasynthesis experiments with A. pretiosum HGF073, a mutant that is unable to produce the essential starter unit AHBA (1) by itself, we achieved the generation of several novel ansamitocin derivatives, among other things, based on the simple mutasynthon 3-amino-5-chlorobenzoic acid (10, Scheme 2).
![[1860-5397-8-96-i2]](/bjoc/content/inline/1860-5397-8-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mutasynthetic preparation of ansamitocin derivatives 11a–h by using 3-amino-5-chlorobenzoic acid (10) as a mutasynthon (yields in % are calculated with reference to the amount of mutasynthon 10 fed).
Scheme 2: Mutasynthetic preparation of ansamitocin derivatives 11a–h by using 3-amino-5-chlorobenzoic acid (10...
With the exception of proansamitocin analogue 11a, all new metabolites were isolated on a preparative scale and fully characterized. These compounds correspond to the known stepwise sequence of postketide synthase tailoring transformations, and those products and similar compounds have been reported by us before [16]. As evident from the variety of compounds isolated, proansamitocin analogues resulting from supplementation of AHBA analogue 10 to A. pretiosum HGF073 were not efficiently processed by the enzymes involved in post-PKS tailoring. In addition to the ordinary compounds 11a–e, the experiment yielded three compounds of an unprecedented type (11f–h) whose appearance could not be attributed to the known tailoring transformations. Herein, we now describe for the first time the isolation and characterization of compounds 11f–h sharing, in contrast to all other (pro)ansamitocin derivatives known so far, the common feature of deoxygenation at C-7. In addition, proansamitocin derivatives 11g–h are notable for their C-9 alcohol, while 11h shows additional carbamoylation of the unusual alcohol moiety. The extraordinary proansamitocin derivatives 11f–h were fully characterized, except for the configuration at C-9 in 11g and 11h (single diastereomer). Overlap with other signals in the 1H NMR spectra hampered complete assignment of all coupling constants at C-9 except for J9,10 = 7.2 Hz, which, however, is not diagnostic.
In continuation of our experiments with advanced biosynthetic intermediates, such as proansamitocin (2) [25] and seco-acid derivatives [26,27] serving as mutasynthons in experiments with early-stage-blocked mutants, we also tested the unusual metabolites 9a and 9b [17] as precursors for further processing by the AHBA(−)-mutant of the ansamitocin producer (Scheme 3).
![[1860-5397-8-96-i3]](/bjoc/content/inline/1860-5397-8-96-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mutasynthetic biotransformation of proansamitocin derivatives 9a and 9b with AHBA(−) mutant A. pretiosum HGF073 (starting materials could be reisolated: 9a: 77% reisolated, 9b: 23% reisolated).
Scheme 3: Mutasynthetic biotransformation of proansamitocin derivatives 9a and 9b with AHBA(−) mutant A. pret...
Originally obtained by fermentation of a mutant blocked to the greatest extent in the post-PKS transformation sequence (A. pretiosum Δasm12/21) [17], it was questionable whether these compounds, differing substantially from proansamitocin (2) both by a rearranged diene system and an alcohol moiety at C-14, would be accepted by the tailoring enzymes. Surprisingly, when the rearranged oxidation products 9a and 9b were each supplemented to cultures of A. pretiosum HGF073, new products were formed. While the carbamoylated derivative 12 resulting from biotransformation of alcohol 9b can be attributed to the activity of the carbamoyltransferase Asm21, thereby providing an indication pertaining to its substrate flexibility, products 13a and 13b obtained from both experiments are more unusual and differ from the starting material by being deoxygenated at C-7.
However, the structure of compound 12 strongly contrasts with all of the other 7-O-carbamoylated (pro)ansamitocin derivatives that we have obtained with this kind of feeding experiment so far [17]. In the case of compound 12, the carbinolamide moiety is not present in a cyclic halfaminal form (δC-9 ~197 ppm instead of ~82 ppm), despite the fact that the keto group at C-9 is still present. This result may be attributed to the keto group at C-9 being in conjugation with the diene system, resulting in a reduced activity of the carbonyl group and an alteration of the macrolactam ring conformation.
We based the determination of deoxygenation at C-7 in 13a and 13b on MS data and the appearance of a secondary carbon atom in exchange of the tertiary carbinol at C-7 on phase-sensitive 1H–13C-correlation NMR spectra (HSQC). It needs to be noted that NOE-analysis combined with molecular modeling did not allow elucidation of the absolute configuration at C-14 for the new derivatives 13a and 13b, as it did not allow for the starting compounds 9a and 9b [17].
In order to shed light on the sequence of events leading to deoxygenation, the carbamoylated derivative 12 was again fed to a culture of A. pretiosum HGF073, and the formation of new products was analyzed by UPLC-HRMS of the partially purified crude extract (Scheme 3). Indeed, the expected deoxygenated product 13b could be detected, but was also accompanied by the formation of alcohol 9b. Compound 9b may either have resulted from Michael addition of water to the suspected intermediate enone 17 (see later in Scheme 5) or from hydrolytic cleavage of the carbinolamide 12.
A blocked mutant of A. pretiosum that is unable to carry out acylation of the C-3 alcohol of the carbamoylated proansamitocin derivative precursor due to genetic inactivation of the acyl transferase Asm19, was reported by Floss et al. [21,28]. It was known that mutant strain A. pretiosum HGF059 produces the expected ansamitocin derivative 14 in good yield (Scheme 4) [28]. Besides the ester side chain, compound 14 also lacks the epoxy functionality and the N-methyl group. These two tailoring steps finalize the biosynthesis of ansamitocin P-3 (AP-3, 4) and occur only after acylation has taken place (Scheme 1).
![[1860-5397-8-96-i4]](/bjoc/content/inline/1860-5397-8-96-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Fermentation products 14–16 of acyl transferase Asm19-blocked mutant A. pretiosum HGF059 (Δasm19) (yields given as isolated product per volume of fermentation broth).
Scheme 4: Fermentation products 14–16 of acyl transferase Asm19-blocked mutant A. pretiosum HGF059 (Δasm19) (...
While examining the fermentation extract for byproducts we were able to identify two new metabolites, 15 and 16. Formation of compound 15 can be traced back to inefficient chlorination, a phenomenon that we have encountered before in other feeding experiments with proansamitocin [25]. More unusual is metabolite 16, which is yet another example of a case in which deoxygenation at C-7 has taken place.
In summary, deoxygenation may occur when the AHBA-blocked mutant A. pretiosum HGF073 or the acyl transferase Asm19-blocked mutant A. pretiosum HGF059 are employed, whereas the reduction process does not occur with the blocked mutant A. pretiosum ∆asm12/21. The major difference between these three mutants is the presence (HGF073, HGF059) or absence (Δasm12/21) of the carbamoyl transferase Asm21. Carbamoylation of proansamitocin derivatives at C-7 (e.g. 11a, 9a/b and 7) introduces a fairly good leaving group β-positioned to the keto group, thereby facilitating elimination to enones 17 (Scheme 5).
![[1860-5397-8-96-i5]](/bjoc/content/inline/1860-5397-8-96-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Possible mechanism of deoxygenation at C-7 of proansamitocin derivatives.
Scheme 5: Possible mechanism of deoxygenation at C-7 of proansamitocin derivatives.
In the case of compound 12, which predominantly exists in a form lacking the typical cyclized carbinolamide moiety, the C-9 keto group can preserve its electronic properties, thus facilitating the elimination step. Finally, the activity of a tailoring reductase, which in all likelihood is not part of the PKS, catalyzes the reduction of the α,β-unsaturated bond, yielding the deoxygenated derivatives (11f, 13a/b, 16). Diastereoselective reduction of the C-9 keto group and carbamoylation of intermediate 11f would then result in compounds 11g and 11h, respectively.
A factor contributing to the formation of deoxygenated proansamitocin derivatives is likely the usage of the carbamoylated precursors by the Asm19 acyl transferase. Acylation is a crucial bottleneck step in the otherwise partly flexible sequence of post-PKS transformations. When this step cannot occur due to the absence of an active acyl transferase (as in A. pretiosum HGF059), or if the transformation is inefficient (e.g., compound 11b) or even nonexistent due to an unusual substrate (e.g., compound 12), carbamoylated intermediates accumulate. Indeed, the carbamoylated product 11b was the major product of the mutasynthesis experiment with mutasynthon 10. The carbamoylated compounds may then be channeled into the pathway leading to deoxygenated products. Depending on the substrate, this process may be quite efficient. For instance, no carbamoylated product could be isolated after the biotransformation of compound 9a, indicating an efficient transformation of the suspected carbamoylated intermediate to the final product 13a.
All (pro)ansamitocin derivatives fully characterized by NMR were also subjected to in vitro biological testing with different human cell lines derived from tumors or the umbilical vein. The results from these tests are given as values for the half-maximal inhibitory concentration of the respective ansamitocin derivatives in comparison to the “gold standard” ansamitocin P-3 (4, Table 1). As expected [14], all (pro)ansamitocin derivatives lacking the ester side chain at C-3 (11b, 11g–h, 12–16) do not show any antiproliferative activity (IC50 > 800 nM) for at least two of the cell lines listed in Table 1.
Table 1: Antiproliferative activity IC50 [nmol/L] of 11c–e in comparison to AP-3 (4).a
| compound | cell line | |||||
|---|---|---|---|---|---|---|
| U-937 | A-431 | SK- OV-3 | PC-3 | MCF-7 | HUVEC | |
| AP-3 (4) | 0.01 | 0.08 | 0.05 | 0.06 | n.d. | 0.02 |
| 11c | 0.5 | 1.58 | 0.66 | 0.30 | 0.90 | 0.32 |
| 11d | 0.05 | 0.10 | 0.05 | 0.16 | 0.11 | 0.08 |
| 11e | 0.18 | 0.35 | 0.21 | 0.53 | 0.41 | 0.21 |
aValues shown are means of two determinations in parallel; human cell lines: U-937 (histiocytic lymphoma), A-431 (epidermoid carcinoma), SK-OV-3 (ovary adenocarcinoma), PC-3 (prostate adenocarcinoma), MCF-7 (breast adenocarcinoma), HUVEC (umbilical vein endothelial cells); n.d. = not determined.
Compounds 11c–e, bearing the ester side chain at C-3, predominantly showed activities in the pM range. As seen also with AP-3 (4), there is no significant difference between cancerous and healthy cells. The most active compound was the N-methyl derivative 11d, which reached the activity of the standard AP-3 (4) for selected cell lines.
Conclusion
In conclusion, we disclose the isolation and chemical and antiproliferative activity characterization of several novel ansamitocin derivatives that are deoxygenated at C-7. We used three different A. pretiosum mutants and a variety of mutasynthetic approaches. These preliminary studies on the deoxygenation at C-7 suggest that it occurs by elimination at C-7/C-8 after carbamoylation has taken place, followed by reduction mediated by an unknown reductase, which is not part of the main assembly line PKS. As expected, all ansamitocin compounds bearing the ester side chain at C-3 predominantly showed activities in the pM range.
Experimental
Analytical details are given in the Supporting Information File 1.
Cultivation
In general, cultivation of microbial strains on agar plates was conducted in a Heraeus incubator at 30 °C, while cultivation in a shake flask was performed in a multilevel New Brunswick Scientific Innova 4900 gyratory multi-shaker at 200 rpm at 29 °C.
Unless otherwise noted, the cultivation media were prepared using distilled water and sterilized by autoclaving: YMG medium – 10 g/L malt extract (Sigma), 4 g/L yeast extract (Bacto), 4 g/L D(+)- glucose∙H2O; YMG agar – YMG medium plus 22 g/L agar (Bacto); K-medium [29], basal composition (final start concentration in the main culture corresponds to 5/6 of the values given for this medium due to dilution) – 60 g/L dextrin from maize starch (Fluka), 30 g/L D(+)-maltose∙H2O (Fluka), 5.25 g/L cottonseed flour (Proflo), 5 g/L CaCO3, 4.5 g/L yeast extract (Bacto), 300 mg/L K2HPO4 (Fluka, TraceSelect), 2 mg/L FeSO4∙7H2O. K-medium, additive – autoclaved and added separately: 3 g/L L-valine (final start concentration in the main culture, from a 3% (w/v) stock solution). A. pretiosum HGF073 is a replicate of strain HGF056 reported in [22].
Fermentation of A. pretiosum strains
A. pretiosum strains (HGF073, HGF059, Δasm12/21) were stored as spore suspensions in 40% (v/v) glycerol/water at −80 °C, and used for the inoculation of YMG agar plates. Following incubation of the plates for 4 d at 30 °C, 5–8 well-sporulated colonies were transferred to a 1.5 mL tube charged with 1 mL of sterile distilled water and filled to approx. 50% height with sterile glass beads (Ø = 2 mm, washed with dilute hydrochloric acid). After vortex-mixing, the resulting suspension was used for the inoculation of precultures in bottom-baffled 250 mL Erlenmeyer flasks charged with YMG medium (50 mL per flask, with additional steel spring). Precultures were shaken for 2 d at 29 °C before inoculation of main production cultures (1/15 dilution). Cultivations were performed in K-medium with additives – 42 mL K-medium with L-valine, 3 mL preculture and one drop of SAG 471 anti-foam (GE Bayer Silicones) [30] – by using nonbaffled 250 mL Erlenmeyer flasks (final volume: 35–60 mL per flask, with additional steel spring). Shaking was continued at 29 °C for a total cultivation time of 7–10 days.
For detection of novel products from test cultures, samples of the culture broth (200 μL) were mixed with ethanol (200 μL), centrifuged (20800g, 3 min, 4 °C) and the clear supernatant subjected to UPLC-ESIMS analysis. Failing detection of novel products, the culture broth was extracted three times with ethyl acetate, dried over MgSO4, concentrated in vacuo, and filtered over silica gel with ethyl acetate, and the solvent was removed in vacuo. The residue was dissolved in methanol (1 mL) and subjected to UPLC-ESIMS analysis.
For isolation of novel products from (large-scale) fermentations, the combined fermentation broth was extracted with ethyl acetate as described above, and the crude extract subjected to a sequence of chromatographic purifications (Supporting Information File 1).
Mutasynthesis with A. pretiosum HGF073
In mutasynthesis experiments with A. pretiosum HGF073, production cultures were shaken for 2 d after inoculation (see above) before mutasynthons were added (9a, 9b, 10 or 12). For novel mutasynthons, productivity of the strain was first monitored by parallel feeding of mutasynthons for which acceptance was known (e.g., the natural starter building block: 3-Amino-5-hydroxybenzoic acid, hydrochloride salt (1)). Mutasynthons were dissolved in DMSO/water [preferably 1:1; volume of feeding solution not exceeding 10% (v/v) with respect to the recipient culture] and sterilized by filtration.
Mutasynthon 10 (1.25 mmol/L of fermentation broth) was added to production cultures with a combined volume of 945 mL continuously (drop-wise) over the time-course of 3.5 d, by using autoclavable, syringe pump-driven feeding capillaries – Braintree Scientific BS-9000-8 syringe pump with Upchurch Scientific high-purity Teflon® PFA tubing (1/16” OD, 0.1” ID) and Tefzel® connectors.
Biotransformation of the proansamitocin derivatives 9a, 9b and 12 was carried out by supplementing a production culture of A. pretiosum HGF073 (45 mL final volume; K-medium; see above) with the respective derivative [9a/b (each: 4.5 mg, 9.8 µmol, dissolved in 2 mL DMSO:H2O = 1:1); 12 (0.1 mg, 0.2 µmol, dissolved in 1 mL DMSO)] in a single portion after 2.5 (for 12) to 3.5 d (for 9a/b) of shaking.
Following biotransformation (refermentation) of proansamitocin derivative 12, only UPLC-ESIMS analysis of the silica-gel-filtered ethyl acetate extract taken up in methanol (see above) was carried out. The retention times and mass spectra of the detected product derivatives 9b and 13b were identical to those of the previously isolated materials (Supporting Information File 1).
Supporting Information
The supporting information provides purification protocols of fermentations, a short description of the cell proliferation assay, analytical descriptions of new metabolites and copies of 1H and 13C NMR spectra.
| Supporting Information File 1: Analytical details and compound spectra. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Cragg, G. M.; Grothaus, P. G.; Newman, D. J. Chem. Rev. 2009, 109, 3012–3043. doi:10.1021/cr900019j
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2007, 70, 461–477. doi:10.1021/np068054v
Return to citation in text: [1] -
von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Angew. Chem. 2006, 118, 5194–5254. doi:10.1002/ange.200600350
Angew. Chem. Int. Ed. 2006, 45, 5072–5129. doi:10.1002/anie.200600350.
Return to citation in text: [1] -
Kirschning, A.; Hahn, F. Angew. Chem. 2012, 124, 4086–4096. doi:10.1002/ange.201107386
Angew. Chem. Int. Ed. 2012, 51, 4012–4022. doi:10.1002/anie.201107386.
Return to citation in text: [1] [2] -
Weist, S.; Süssmuth, R. D. Appl. Microbiol. Biotechnol. 2005, 68, 141–150. doi:10.1007/s00253-005-1891-8
Return to citation in text: [1] -
Kirschning, A.; Taft, F.; Knobloch, T. Org. Biomol. Chem. 2007, 5, 3245–3295. doi:10.1039/b709549j
Return to citation in text: [1] -
Kennedy, J. Nat. Prod. Rep. 2008, 25, 25–34. doi:10.1039/B707678A
Return to citation in text: [1] -
Funayama, S.; Cordell, G. A. In Studies in Natural Product Chemistry; Rahman, A.-U., Ed.; Elsevier Science B. V.: Amsterdam, 2000; Vol. 23, pp 51–106.
Return to citation in text: [1] -
Hamel, E. Pharmacol. Ther. 1992, 55, 31–51. doi:10.1016/0163-7258(92)90028-X
Return to citation in text: [1] -
Cassady, J. M.; Chan, K. K.; Floss, H. G.; Leistner, E. Chem. Pharm. Bull. 2004, 52, 1–26. doi:10.1248/cpb.52.1
Return to citation in text: [1] -
Kirschning, A.; Harmrolfs, K.; Knobloch, T. C. R. Chim. 2008, 11, 1523–1543. doi:10.1016/j.crci.2008.02.006
Return to citation in text: [1] -
Floss, H. G.; Yu, T.-W.; Arakawa, K. J. Antibiot. 2011, 64, 35–44. doi:10.1038/ja.2010.139
Return to citation in text: [1] -
Taft, F.; Brünjes, M.; Floss, H. G.; Czempinski, N.; Grond, S.; Sasse, F.; Kirschning, A. ChemBioChem 2008, 9, 1057–1060. doi:10.1002/cbic.200700742
Return to citation in text: [1] [2] -
Kubota, T.; Brünjes, M.; Frenzel, T.; Xu, J.; Kirschning, A.; Floss, H. G. ChemBioChem 2006, 7, 1221–1225. doi:10.1002/cbic.200500506
Return to citation in text: [1] [2] [3] -
Taft, F.; Brünjes, M.; Knobloch, T.; Floss, H. G.; Kirschning, A. J. Am. Chem. Soc. 2009, 131, 3812–3813. doi:10.1021/ja8088923
Return to citation in text: [1] [2] -
Knobloch, T.; Harmrolfs, K.; Taft, F.; Thomaszewski, B.; Sasse, F.; Kirschning, A. ChemBioChem 2011, 12, 540–547. doi:10.1002/cbic.201000608
Return to citation in text: [1] [2] [3] [4] -
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Eichner, S.; Floss, H. G.; Sasse, F.; Kirschning, A. ChemBioChem 2009, 10, 1801–1805. doi:10.1002/cbic.200900246
Return to citation in text: [1] -
Eichner, S.; Eichner, T.; Floss, H. G.; Fohrer, J.; Hofer, E.; Sasse, F.; Zeilinger, C.; Kirschning, A. J. Am. Chem. Soc. 2012, 134, 1673–1679. doi:10.1021/ja2087147
Return to citation in text: [1] -
Walsh, C. T.; Haynes, S. W.; Ames, B. D. Nat. Prod. Rep. 2012, 29, 37–59. doi:10.1039/c1np00072a
Return to citation in text: [1] [2] -
Spiteller, P.; Bai, L.; Shang, G.; Carroll, B. J.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2003, 125, 14236–14237. doi:10.1021/ja038166y
Return to citation in text: [1] [2] [3] -
Yu, T.-W.; Bai, L.; Clade, D.; Hoffmann, D.; Toelzer, S.; Trinh, K. Q.; Xu, J.; Moss, S. J.; Leistner, E.; Floss, H. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7968–7973. doi:10.1073/pnas.092697199
Return to citation in text: [1] [2] [3] -
Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 10644–10645. doi:10.1021/ja0206339
Return to citation in text: [1] [2] -
Yu, T.-W.; Müller, R.; Müller, M.; Zhang, X.; Dräger, G.; Kim, C.-G.; Leistner, E.; Floss, H. G. J. Biol. Chem. 2001, 276, 12546–12555. doi:10.1074/jbc.M009667200
Return to citation in text: [1] [2] -
Meyer, A.; Brünjes, M.; Taft, F.; Frenzel, T.; Sasse, F.; Kirschning, A. Org. Lett. 2007, 9, 1489–1492. doi:10.1021/ol0702270
Return to citation in text: [1] [2] -
Frenzel, T.; Brünjes, M.; Quitschalle, M.; Kirschning, A. Org. Lett. 2006, 8, 135–138. doi:10.1021/ol052588q
Return to citation in text: [1] -
Harmrolfs, K.; Brünjes, M.; Dräger, G.; Floss, H. G.; Sasse, F.; Taft, F.; Kirschning, A. ChemBioChem 2010, 11, 2517–2520. doi:10.1002/cbic.201000422
Return to citation in text: [1] -
Moss, S. J.; Bai, L.; Toelzer, S.; Caroll, B. J.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 6544–6545. doi:10.1021/ja020214b
Return to citation in text: [1] [2] -
Bandi, S.; Kim, Y. J.; Sa, S. O.; Chang, Y.-K. J. Microbiol. Biotechnol. 2005, 15, 930–937.
Return to citation in text: [1] -
Chung, J.; Byng, G. S. Mutant Actinosynnema pretiosum strain with increased maytansinoid production. U.S. Patent 6,790,954, Sept 14, 2004.
Use of SAG 471 anti-foam for fermentation of A. pretiosum was suggested.
Return to citation in text: [1]
| 14. | Kubota, T.; Brünjes, M.; Frenzel, T.; Xu, J.; Kirschning, A.; Floss, H. G. ChemBioChem 2006, 7, 1221–1225. doi:10.1002/cbic.200500506 |
| 29. | Bandi, S.; Kim, Y. J.; Sa, S. O.; Chang, Y.-K. J. Microbiol. Biotechnol. 2005, 15, 930–937. |
| 22. | Yu, T.-W.; Bai, L.; Clade, D.; Hoffmann, D.; Toelzer, S.; Trinh, K. Q.; Xu, J.; Moss, S. J.; Leistner, E.; Floss, H. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7968–7973. doi:10.1073/pnas.092697199 |
| 1. | Cragg, G. M.; Grothaus, P. G.; Newman, D. J. Chem. Rev. 2009, 109, 3012–3043. doi:10.1021/cr900019j |
| 2. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2007, 70, 461–477. doi:10.1021/np068054v |
| 3. |
von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Angew. Chem. 2006, 118, 5194–5254. doi:10.1002/ange.200600350
Angew. Chem. Int. Ed. 2006, 45, 5072–5129. doi:10.1002/anie.200600350. |
| 8. | Funayama, S.; Cordell, G. A. In Studies in Natural Product Chemistry; Rahman, A.-U., Ed.; Elsevier Science B. V.: Amsterdam, 2000; Vol. 23, pp 51–106. |
| 13. | Taft, F.; Brünjes, M.; Floss, H. G.; Czempinski, N.; Grond, S.; Sasse, F.; Kirschning, A. ChemBioChem 2008, 9, 1057–1060. doi:10.1002/cbic.200700742 |
| 14. | Kubota, T.; Brünjes, M.; Frenzel, T.; Xu, J.; Kirschning, A.; Floss, H. G. ChemBioChem 2006, 7, 1221–1225. doi:10.1002/cbic.200500506 |
| 15. | Taft, F.; Brünjes, M.; Knobloch, T.; Floss, H. G.; Kirschning, A. J. Am. Chem. Soc. 2009, 131, 3812–3813. doi:10.1021/ja8088923 |
| 16. | Knobloch, T.; Harmrolfs, K.; Taft, F.; Thomaszewski, B.; Sasse, F.; Kirschning, A. ChemBioChem 2011, 12, 540–547. doi:10.1002/cbic.201000608 |
| 4. |
Kirschning, A.; Hahn, F. Angew. Chem. 2012, 124, 4086–4096. doi:10.1002/ange.201107386
Angew. Chem. Int. Ed. 2012, 51, 4012–4022. doi:10.1002/anie.201107386. |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 5. | Weist, S.; Süssmuth, R. D. Appl. Microbiol. Biotechnol. 2005, 68, 141–150. doi:10.1007/s00253-005-1891-8 |
| 6. | Kirschning, A.; Taft, F.; Knobloch, T. Org. Biomol. Chem. 2007, 5, 3245–3295. doi:10.1039/b709549j |
| 7. | Kennedy, J. Nat. Prod. Rep. 2008, 25, 25–34. doi:10.1039/B707678A |
| 21. | Spiteller, P.; Bai, L.; Shang, G.; Carroll, B. J.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2003, 125, 14236–14237. doi:10.1021/ja038166y |
| 22. | Yu, T.-W.; Bai, L.; Clade, D.; Hoffmann, D.; Toelzer, S.; Trinh, K. Q.; Xu, J.; Moss, S. J.; Leistner, E.; Floss, H. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7968–7973. doi:10.1073/pnas.092697199 |
| 23. | Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 10644–10645. doi:10.1021/ja0206339 |
| 24. | Yu, T.-W.; Müller, R.; Müller, M.; Zhang, X.; Dräger, G.; Kim, C.-G.; Leistner, E.; Floss, H. G. J. Biol. Chem. 2001, 276, 12546–12555. doi:10.1074/jbc.M009667200 |
| 4. |
Kirschning, A.; Hahn, F. Angew. Chem. 2012, 124, 4086–4096. doi:10.1002/ange.201107386
Angew. Chem. Int. Ed. 2012, 51, 4012–4022. doi:10.1002/anie.201107386. |
| 16. | Knobloch, T.; Harmrolfs, K.; Taft, F.; Thomaszewski, B.; Sasse, F.; Kirschning, A. ChemBioChem 2011, 12, 540–547. doi:10.1002/cbic.201000608 |
| 18. | Eichner, S.; Floss, H. G.; Sasse, F.; Kirschning, A. ChemBioChem 2009, 10, 1801–1805. doi:10.1002/cbic.200900246 |
| 19. | Eichner, S.; Eichner, T.; Floss, H. G.; Fohrer, J.; Hofer, E.; Sasse, F.; Zeilinger, C.; Kirschning, A. J. Am. Chem. Soc. 2012, 134, 1673–1679. doi:10.1021/ja2087147 |
| 21. | Spiteller, P.; Bai, L.; Shang, G.; Carroll, B. J.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2003, 125, 14236–14237. doi:10.1021/ja038166y |
| 22. | Yu, T.-W.; Bai, L.; Clade, D.; Hoffmann, D.; Toelzer, S.; Trinh, K. Q.; Xu, J.; Moss, S. J.; Leistner, E.; Floss, H. G. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7968–7973. doi:10.1073/pnas.092697199 |
| 23. | Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 10644–10645. doi:10.1021/ja0206339 |
| 24. | Yu, T.-W.; Müller, R.; Müller, M.; Zhang, X.; Dräger, G.; Kim, C.-G.; Leistner, E.; Floss, H. G. J. Biol. Chem. 2001, 276, 12546–12555. doi:10.1074/jbc.M009667200 |
| 13. | Taft, F.; Brünjes, M.; Floss, H. G.; Czempinski, N.; Grond, S.; Sasse, F.; Kirschning, A. ChemBioChem 2008, 9, 1057–1060. doi:10.1002/cbic.200700742 |
| 14. | Kubota, T.; Brünjes, M.; Frenzel, T.; Xu, J.; Kirschning, A.; Floss, H. G. ChemBioChem 2006, 7, 1221–1225. doi:10.1002/cbic.200500506 |
| 15. | Taft, F.; Brünjes, M.; Knobloch, T.; Floss, H. G.; Kirschning, A. J. Am. Chem. Soc. 2009, 131, 3812–3813. doi:10.1021/ja8088923 |
| 16. | Knobloch, T.; Harmrolfs, K.; Taft, F.; Thomaszewski, B.; Sasse, F.; Kirschning, A. ChemBioChem 2011, 12, 540–547. doi:10.1002/cbic.201000608 |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 20. | Walsh, C. T.; Haynes, S. W.; Ames, B. D. Nat. Prod. Rep. 2012, 29, 37–59. doi:10.1039/c1np00072a |
| 10. | Cassady, J. M.; Chan, K. K.; Floss, H. G.; Leistner, E. Chem. Pharm. Bull. 2004, 52, 1–26. doi:10.1248/cpb.52.1 |
| 11. | Kirschning, A.; Harmrolfs, K.; Knobloch, T. C. R. Chim. 2008, 11, 1523–1543. doi:10.1016/j.crci.2008.02.006 |
| 12. | Floss, H. G.; Yu, T.-W.; Arakawa, K. J. Antibiot. 2011, 64, 35–44. doi:10.1038/ja.2010.139 |
| 30. |
Chung, J.; Byng, G. S. Mutant Actinosynnema pretiosum strain with increased maytansinoid production. U.S. Patent 6,790,954, Sept 14, 2004.
Use of SAG 471 anti-foam for fermentation of A. pretiosum was suggested. |
| 20. | Walsh, C. T.; Haynes, S. W.; Ames, B. D. Nat. Prod. Rep. 2012, 29, 37–59. doi:10.1039/c1np00072a |
| 25. | Meyer, A.; Brünjes, M.; Taft, F.; Frenzel, T.; Sasse, F.; Kirschning, A. Org. Lett. 2007, 9, 1489–1492. doi:10.1021/ol0702270 |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 16. | Knobloch, T.; Harmrolfs, K.; Taft, F.; Thomaszewski, B.; Sasse, F.; Kirschning, A. ChemBioChem 2011, 12, 540–547. doi:10.1002/cbic.201000608 |
| 28. | Moss, S. J.; Bai, L.; Toelzer, S.; Caroll, B. J.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 6544–6545. doi:10.1021/ja020214b |
| 25. | Meyer, A.; Brünjes, M.; Taft, F.; Frenzel, T.; Sasse, F.; Kirschning, A. Org. Lett. 2007, 9, 1489–1492. doi:10.1021/ol0702270 |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 21. | Spiteller, P.; Bai, L.; Shang, G.; Carroll, B. J.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2003, 125, 14236–14237. doi:10.1021/ja038166y |
| 28. | Moss, S. J.; Bai, L.; Toelzer, S.; Caroll, B. J.; Mahmud, T.; Yu, T.-W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 6544–6545. doi:10.1021/ja020214b |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
| 26. | Frenzel, T.; Brünjes, M.; Quitschalle, M.; Kirschning, A. Org. Lett. 2006, 8, 135–138. doi:10.1021/ol052588q |
| 27. | Harmrolfs, K.; Brünjes, M.; Dräger, G.; Floss, H. G.; Sasse, F.; Taft, F.; Kirschning, A. ChemBioChem 2010, 11, 2517–2520. doi:10.1002/cbic.201000422 |
| 17. |
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem. 2012, 124, 776–781. doi:10.1002/ange.201106249
Angew. Chem. Int. Ed. 2012, 51, 752–757. doi:10.1002/anie.201106249. |
© 2012 Knobloch et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)