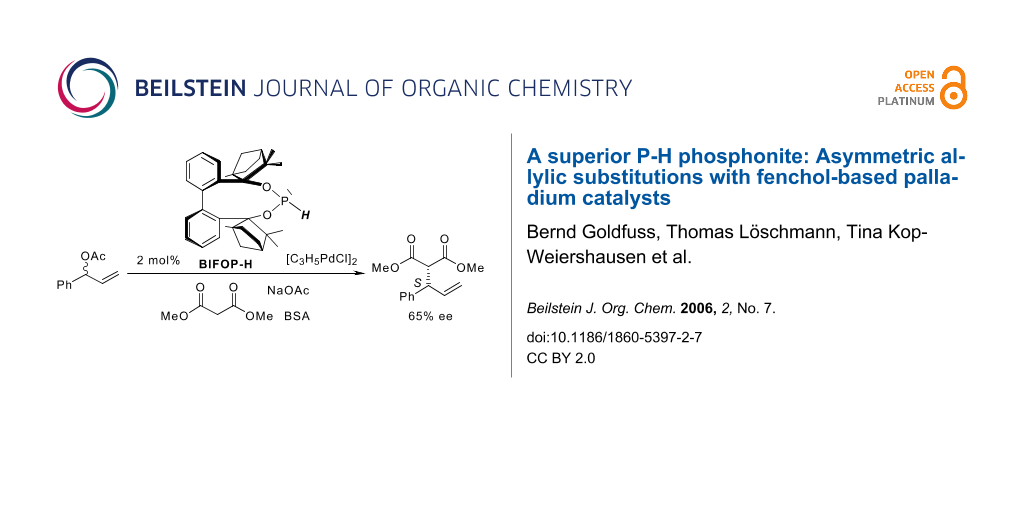
Abstract
The fenchol-based P-H phosphonite BIFOP-H exeeds with 65% ee other monodentate ligands in the Pd-catalyzed substitution of 1-phenyl-2-propenyl acetate with dimethylmalonate.

Graphical Abstract
Introduction
Palladium catalyzed allylic substitutions provide valuable tools for stereoselective C-C- and C-heteroatom connections [1,2]. The control of regio- and enantioselectivity is challenging, especially with unsymmetrical substrates, e.g. with monoaryl allyl units. According to computational analyses of electronic effects,[3,4] regioselectivity in favor of the branched product is supported at strong donor-substituted (e.g. alkyl, O-alkyl) allylic positions. Frequently employed Pd-catalysts most often favor linear, nonchiral products (Scheme 1).
![[1860-5397-2-7-i1]](/bjoc/content/inline/1860-5397-2-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pd-catalyzed allylic substitution with unsymmetrical substrates (Nu = dimethylmalonate, Nf = OAc).
Scheme 1: Pd-catalyzed allylic substitution with unsymmetrical substrates (Nu = dimethylmalonate, Nf = OAc).
Pfaltz et al. improved the yield of the chiral, branched product by employing electron withdrawing substituents on the P-donor atoms in P, N-oxazoline ligands[5] (Scheme 2) [6]. Such phosphites were thought to favor a more SN1-like addition at the substituted, allylic C-atom. High regio- and enantioselectivities were also achieved with biphenylphosphites by Pamies et al. (Scheme 2) [7].
![[1860-5397-2-7-i2]](/bjoc/content/inline/1860-5397-2-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Bidentate P, N-ligands and a monodentate phosphoramidite for Pd-catalyzed allylic substitutions with unsymmetric substrates, cf. Scheme 1.
Scheme 2: Bidentate P, N-ligands and a monodentate phosphoramidite for Pd-catalyzed allylic substitutions wit...
Besides bidentate P, N-ligands, monodentate ligands are useful, as was demonstrated successfully by Hayashi et al. with the MeO-MOP ligand, yielding 90% branched product with 87% ee for a C-methylated malonate nucleophile and the 4-methoxyphenylallyl substrate [8]. Van Leeuwen's bulky, monodentate TADDOL based phosphoramidite gave rise to intriguing memory effects [28b] and yielded 6% branched product with 25% ee (Scheme 2) [9].
We have recently employed modular, chelating fencholates, [10-14] in enantioselective organozinc catalysts, [15-19] and in chiral n-butyllithium aggregates [20-24]. In Pd-catalyzed allylic substitutions of diphenylallyl acetate, fenchyl diphenylphosphinites (FENOPs) with phenyl or anisyl groups favor the S-enantiomer, but with a 2-pyridyl unit the R-enantiomer was preferred (Scheme 3).[25] According to computational transition structure analyses, these phenyl and anisyl phosphinites are not "monodentate" but form chelate complexes via π-coordination. Biphenyl-2,2'-bisfenchol (BIFOL)[13] was developed as combination of a flexible biaryl axis (as in BINOL) and sterically crowded hydroxy groups (as in TADDOLs). BIFOL based phosphanes (BIFOPs) are sterically highly hindered and were employed in copper-catalyzed 1,4-additions of diethylzinc to 2-cyclohexenone [26].
![[1860-5397-2-7-i3]](/bjoc/content/inline/1860-5397-2-7-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Fenchole-based phosphorus ligands (i.e. FEENOPs and BIFOPs) for Pd-catalyzed allylic substitutions. Pd-π arene or Pd-N coordinations give rise to different enantioselectivitites.
Scheme 3: Fenchole-based phosphorus ligands (i.e. FEENOPs and BIFOPs) for Pd-catalyzed allylic substitutions....
Here we use a selection of fenchol-based bidentate pyridine FENOP- and monodentate BIFOP-ligands in Pd-catalysts to study allylic substitutions of the challenging 1-phenyl-2-propenyl acetate (Scheme 1, R=Ph) [27,28].
Results and discussion
Fenchylphosphinites (FENOPs) and biphenylbisfenchol based phosphorus ligands are all suitable for Pd-catalyzed allylic alkylations of 1-phenyl-2-propenyl acetate (Scheme 4, Table 1, see Supporting Information File 1 for full experimental data).
![[1860-5397-2-7-i4]](/bjoc/content/inline/1860-5397-2-7-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Allylic alkylation of 1-phenyl-2-propenyl acetate by sodium dimethylmalonate (BSA-method) with Pd-FENOP- or Pd-BIFOP- catalysts.
Scheme 4: Allylic alkylation of 1-phenyl-2-propenyl acetate by sodium dimethylmalonate (BSA-method) with Pd-F...
Table 1: FENOP- and BIFOP-Pd-catalysts in enantioselective allylic substitutions of phenylallyacetate by dimethylmalonate.a)
| Ligand | Linear / branched b) | % ee (major enantiomer) c) | % yield b) |
|---|---|---|---|
| FENOP | 42 / 58 | 19 (R) | 54 |
| FENOP-Me | 39 / 61 | 31 (R) | 43 |
| FENOP-NMe2 | 44 / 56 | 37 (R) | 50 |
| BIFOP-Cl | 89 / 11 | 39 (S) | 60 |
| BIFOP-Br | 85 / 15 | 37 (S) | 56 |
| BIFOP-H | 80 / 20 | 65 (S) | 68 |
| BIFOP-Et | 85 / 15 | 8 (S) | 70 |
| BIFOP-nBu | 65 / 35 | 5 (S) | 75 |
| BIFOP-Oph | 68 / 32 | 29 (S) | 58 |
| BIFOP-NEt2 | 52 / 48 | 10 (S) | 52 |
a) All catalyses were performed in THF, 12 h at -78°C then 24 h at RT with 0.0055 mmol of the ligand, 0.0055 mmol of [Pd(allyl)Cl]2 (1 mol% catalyst) and 0.57 mol of 1-phenylallylacetate substrate.
b) Linear / branched ratios as well as yields were determined by integration of 1H-NMR spectra.
c) Enantiomeric excesses (%ee) of the branched products were determined by HPLC (Daicel-OD-H, hexanes / i-PrOH = 99/1, 0.55 mi /min., l= 220 nm, tR= 16.7 min. (R), 17.7 min. (S).
All three P, N-bidentate FENOP ligands, FENOP, FENOP-Me and FENOP-NMe2, favor branched alkylation products (Table 1). This tendency towards formation of chiral, branched products is even apparent from X-ray crystal structure analyses of corresponding Pd-phenylallyl intermediates. All three Pd-allyl complexes, Pd-FENOP, Pd-FENOP-Me and Pd-FENOP-NMe2 (Figure 1, Figure 2 and Figure 3) exhibit the allylic phenyl group trans situated relative to phosphorus. Rather long C3-Pd distances (2.30 Å, 2.30 Å and 2.25 Å) are apparent for these trans position in comparison to the shorter C1-Pd bond distances (2.13 Å, 2.08 Å and 2.13 Å, cf. Figure 1, Figure 2 and Figure 3). This differentiation agrees with the "trans to phosphorus" rule, [1,29,30] which predicts the attack of the nucleophile (i.e. malonate) at the weakest (longest) C3-Pd bond, yielding preferably the chiral, branched product.
![[1860-5397-2-7-1]](/bjoc/content/figures/1860-5397-2-7-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of the cationic complex Pd-FENOP (CCDC 299944), the perchlorate counterion and hydrogen atoms are omitted. The allylic phenyl groups is positioned trans to phosphorus. In agreement with the the "trans rule", C3-Pd is longer then C1-Pd. The nucleophile (i.e. malonate) is expected to attack at C3 yielding the branched product. Distances are given in Angstroms.
Figure 1: X-ray crystal structure of the cationic complex Pd-FENOP (CCDC 299944), the perchlorate counterion ...
![[1860-5397-2-7-2]](/bjoc/content/figures/1860-5397-2-7-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of the cationic complex Pd-FENOP-Me (CCDC 600369), the perchlorate counterion and hydrogen atoms are omitted. The allylic phenyl groups is positioned trans to phosphorus. In agreement with the "trans rule", C3-Pd is longer then C1-Pd. The nucleophile (i.e. malonate) is expected to attack at C3 yielding the branched product. Distances are given in Angstroms.
Figure 2: X-ray crystal structure of the cationic complex Pd-FENOP-Me (CCDC 600369), the perchlorate counteri...
![[1860-5397-2-7-3]](/bjoc/content/figures/1860-5397-2-7-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of the cationic complex Pd-FENOP-NMe2 (CCDC 600370), the perchlorate counterion and hydrogen atoms are omitted. The allylic phenyl groups is positioned trans to phosphorus. In agreement with the the "trans rule", C3-Pd is longer then C1-Pd. The nucleophile (i.e. malonate) is expected to attack at C3 yielding the branched product. The mean values of two independent complexes are given, distances are given in Angstroms.
Figure 3: X-ray crystal structure of the cationic complex Pd-FENOP-NMe2 (CCDC 600370), the perchlorate counte...
Monodentate BIFOP ligands yield more of the linear alkylation product (Table 1), despite their huge steric demand. Surprisingly, the chloro- and bromophosphites, BIFOP-Cl and BIFOP-Br, are stable ligands under these reaction conditions: no conversion with nucleophiles (e.g. malonate), as was observed previously with diethylzinc,[26] was found. The ligands were recovered after catalysis. Apparently, the absence of strongly Lewis-acidic electrophiles (Na+ vs. Zn2+) and the huge steric shielding prevents halide substitutions and BIFOP-Cl(Br) decompositions.
With regard to enantioselectivities, some monodentate BIFOPs are even superior to the pyridine-phosphinites (FENOPs). While FENOPs favor the R-enantiomeric product, the S-enantiomer is preferred by all BIFOP ligands. Enantioselectivities increase from FENOP with 19% ee to FENOP-Me with 31% ee and to FENOP-NMe2 with 37% ee, reflecting the effect of steric demanding and electron donating pyridine groups on enantioselectivity.
The surprisingly stable halogen phosphites BIFOP-Cl and BIFOP-Br yield even higher enantioselectivities (39% and 37% ee) than the corresponding phosphite BIFOP-OPh or the phosphoramidite BIFOP-NEt2 (10% and 29% ee, Table 1). To our knowledge, this is the first successful application of halogen phosphites as ligands in enantioselective catalysis [26]. The highest enantioselectivity however is achieved with the P-H phosphonite BIFOP-H (65% ee, Table 1). As in copper-catalyzed 1,4-additions of diethylzinc to cyclohexenone,[26] the small steric hindrance of the hydrido-substituent and the shielding by the chiral bis-fenchane cavity provide the best combination among the tested BIFOPs for the P-H phosphonite BIFOP-H.
Computational transition structure analyses of allylic substitutions with ammonia mimicking the malonate nucleophile help to understand origins of enantioselectivities,[31-34] as we have shown recently for Pd-FENOP catalysts with the diphenyl allyl substrate [25]. For the P, N-bidentate pyridyl FENOP system, an exo allyl arrangement and a trans to phosphorus addition of the nukleophile is slightly preferred (cf. the two most stable transition state in Figure 4). This favored Si-addition of the nucleophile explains the experimentally observed formation of the R-alkylation product (Table 1). Systematic conformational analyses of transition structures with BIFOP-H in allylic substitutions yields BIFOP-H-Re as the most stable transition structure. Its Re-addition of the NH3-nucleophile is slightly more favored than the Si-addition in the competing transition structure BIFOP-H-Si (Figure 5). This agrees with the experimentally observed formation of the S-alkylation product with BIFOP-ligands (Table 1).
![[1860-5397-2-7-4]](/bjoc/content/figures/1860-5397-2-7-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The two most stable ONIOM(B3LYP/SDD(+ECP) (Pd) /6-31G* (C, H, O, N, P) : UFF) optimized transition structures with FENOP. ZPE (unscaled) corrected total extrapolated energies: FENOP-exo-N (re): -1236.56193 H, FENOP-exo-P (si): -1236.56221 H. The by 0.2 kcal mol-1 slightly preferred si-addition of the NH3 model nucleophile corresponds to the experimental R-alkylation product.
Figure 4: The two most stable ONIOM(B3LYP/SDD(+ECP) (Pd) /6-31G* (C, H, O, N, P) : UFF) optimized transition ...
![[1860-5397-2-7-5]](/bjoc/content/figures/1860-5397-2-7-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The two most stable ONIOM(B3LYP/SDD(+ECP) (Pd) /6-31G* (C, H, O, N, P) : UFF) optimized transition structures with BIFOP-H, due to systematic conformational analysis (60° rotations at P-Pd). ZPE (unscaled) corrected total extrapolated energies: BIFOP-H-re: -1025.01553 H, BIFOP-H-si: -1025.01466 H. The by 0.5 kcal mol-1 slightly preferred re-addition of the NH3 model nucleophile corresponds to the experimental S-alkylation product.
Figure 5: The two most stable ONIOM(B3LYP/SDD(+ECP) (Pd) /6-31G* (C, H, O, N, P) : UFF) optimized transition ...
Conclusion
Besides P, N-bidentate FENOP ligands, monodentate BIFOP ligands can be employed successfully in Pd-catalyzed allylic substitution of 1-phenyl-2-propenyl acetate with dimethylmalonate. Surprisingly, the halogen phosphites BIFOP-Cl and BIFOP-Br are stable towards nucleophiles under catalysis conditions, apparently due to absence of strongly Lewis-acidic cations and the large steric shielding of the phosphorus-halogen functions. With respect to enantioselectivities, the P-H phosphonite BIFOP-H is clearly superior and reaches 65% ee, a rather high selectivity for a monodentate ligand.
Supporting Information
| Supporting Information File 1: contains all experimental data | ||
| Format: PDF | Size: 83.1 KB | Download |
Acknowledgements
We are grateful to the Fonds der Chemischen Industrie for financial support as well as for a Dozenten-Stipendium to B.G. We especially thank the Deutsche Forschungsgemeinschaft (DFG) for funding (GO-930/9, GO-930/7 and GO-930/5) as well as the Bayer AG, the BASF AG, the Wacker AG, the Degussa AG, the Raschig GmbH, the Symrise GmbH, the Solvay GmbH and the OMG AG for generous support.
References
-
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi:10.1021/cr020027w
Return to citation in text: [1] [2] -
Weihofen, R.; Dahnz, A.; Tverskoy, O.; Helmchen, G. Chem. Commun. 2005, 3541. doi:10.1039/b505197e
Return to citation in text: [1] -
Delbecq, F.; Lapouge, C. Organometallics 2000, 19, 2716. doi:10.1021/om0003032
Return to citation in text: [1] -
Goldfuss, B. J. Organomet. Chem. 2006, 691, 4508–4513. doi:10.1016/j.jorganchem.2006.01.061
Return to citation in text: [1] -
Sprinz, J.; Helmchen, G. Tetrahedron Lett. 1993, 34, 1769. doi:10.1016/S0040-4039(00)60774-8
Return to citation in text: [1] -
Pretot, R.; Pfaltz, A. Angew. Chem. 1998, 110, 337.
Angew. Chem., Int. Ed. 1998, 37, 323. doi:10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T
Return to citation in text: [1] -
Pamies, O.; Dieguez, M.; Claver, C. J. Am. Chem. Soc. 2005, 127, 3646. doi:10.1021/ja0425738
Return to citation in text: [1] -
Hayashi, T. J. Organomet. Chem. 1999, 576, 195. doi:10.1016/S0022-328X(98)01058-4
Return to citation in text: [1] -
Boele, M. D. K.; Kamer, P. C. J.; Lutz, M.; Spek, A. L.; de Vries, J. G.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Chem.–Eur. J. 2004, 10, 6232. doi:10.1002/chem.200400154
Return to citation in text: [1] -
Lange, D.; Neudörfl, J.; Goldfuss, B. Tetrahedron 2006, 62, 3704. doi:10.1016/j.tet.2006.01.060
Return to citation in text: [1] -
Soki, F.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2005, 61, 10449. doi:10.1016/j.tet.2005.08.089
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2001, 7, 2028. doi:10.1002/1521-3765(20010504)7:9<2028::AID-CHEM2028>3.0.CO;2-Y
Return to citation in text: [1] -
Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881. doi:10.1016/S0040-4020(99)01077-7
Return to citation in text: [1] [2] -
Goldfuss, B.; Eisenträger, F. Aust. J. Chem. 2000, 53, 209. doi:10.1071/CH99184
Return to citation in text: [1] -
Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166. doi:10.1007/s0089400060166
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77. doi:10.1021/jo991070v
Return to citation in text: [1] -
Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927. doi:10.1021/om990184u
Return to citation in text: [1] -
Goldfuss, B. Synthesis 2005, 2271. doi:10.1055/s-2005-872107
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem.–Eur. J. 2005, 11, 4019. doi:10.1002/chem.200500158
Return to citation in text: [1] -
Goldfuss, B. Enantioselective addition of organolithiums to C=O groups. In Organolithiums in Enantioselective Synthesis; Hodgson, D. M., Ed.; Topics in Organometallic Chemistry, Vol. 5; Springer: Berlin, 2003; pp 21–35.
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem.–Eur. J. 2001, 7, 4456. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S
Return to citation in text: [1] -
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299.
Angew. Chem., Int. Ed. 2000, 39, 4133. doi:10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X
Return to citation in text: [1] -
Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273
Return to citation in text: [1] [2] -
Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6
Return to citation in text: [1] [2] [3] [4] -
Löschmann, T. Chirale P/N-Liganden: Fenchol-Derivate für enantioselektive Katalysen. Ph.D. Thesis, University of Heidelberg, Heidelberg, 2003.
Return to citation in text: [1] -
Kop-Weiershausen, T. Biphenyl-Bisfenchol-Phosphan-Systeme (BIFOPs): Synthesen, strukturelle Studien & Einsatz in enantioselektiver Katalyse. Ph.D. Thesis, University of Cologne, Cologne, 2006.
Return to citation in text: [1] -
Steinhagen, H.; Reggelin, M.; Helmchen, G. Angew. Chem. 1997, 109, 2199.
Angew. Chem., Int. Ed. 1997, 36, 2108. doi:10.1002/anie.199721081
Return to citation in text: [1] -
Goldfuss, B.; Kazmaier, U. Tetrahedron 2000, 56, 6493. doi:10.1016/S0040-4020(00)00613-X
The "trans phosphane preference" also explains memory effects in allylic substitutions.
Return to citation in text: [1] -
Kollmar, M.; Steinhagen, H.; Janssen, J. P.; Goldfuss, B.; Malinovskaya, S. A.; Vázquez, J.; Rominger, F.; Helmchen, G. Chem.–Eur. J. 2002, 8, 3103. doi:10.1002/1521-3765(20020715)8:14<3103::AID-CHEM3103>3.0.CO;2-C
Return to citation in text: [1] -
Vázquez, J.; Goldfuss, B.; Helmchen, G. J. Organomet. Chem. 2002, 641, 67. doi:10.1016/S0022-328X(01)01308-0
Return to citation in text: [1] -
Kollmar, M.; Goldfuss, B.; Reggelin, M.; Rominger, F.; Helmchen, G. Chem.–Eur. J. 2001, 7, 4913. doi:10.1002/1521-3765(20011119)7:22<4913::AID-CHEM4913>3.0.CO;2-7
Return to citation in text: [1] -
Hagelin, H.; Akermark, B.; Norrby, P.-O. Chem.–Eur. J. 1999, 5, 902. doi:10.1002/(SICI)1521-3765(19990301)5:3<902::AID-CHEM902>3.0.CO;2-W
Return to citation in text: [1]
| 31. | Kollmar, M.; Steinhagen, H.; Janssen, J. P.; Goldfuss, B.; Malinovskaya, S. A.; Vázquez, J.; Rominger, F.; Helmchen, G. Chem.–Eur. J. 2002, 8, 3103. doi:10.1002/1521-3765(20020715)8:14<3103::AID-CHEM3103>3.0.CO;2-C |
| 32. | Vázquez, J.; Goldfuss, B.; Helmchen, G. J. Organomet. Chem. 2002, 641, 67. doi:10.1016/S0022-328X(01)01308-0 |
| 33. | Kollmar, M.; Goldfuss, B.; Reggelin, M.; Rominger, F.; Helmchen, G. Chem.–Eur. J. 2001, 7, 4913. doi:10.1002/1521-3765(20011119)7:22<4913::AID-CHEM4913>3.0.CO;2-7 |
| 34. | Hagelin, H.; Akermark, B.; Norrby, P.-O. Chem.–Eur. J. 1999, 5, 902. doi:10.1002/(SICI)1521-3765(19990301)5:3<902::AID-CHEM902>3.0.CO;2-W |
| 26. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 26. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 1. | Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi:10.1021/cr020027w |
| 2. | Weihofen, R.; Dahnz, A.; Tverskoy, O.; Helmchen, G. Chem. Commun. 2005, 3541. doi:10.1039/b505197e |
| 7. | Pamies, O.; Dieguez, M.; Claver, C. J. Am. Chem. Soc. 2005, 127, 3646. doi:10.1021/ja0425738 |
| 1. | Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi:10.1021/cr020027w |
| 29. |
Steinhagen, H.; Reggelin, M.; Helmchen, G. Angew. Chem. 1997, 109, 2199.
Angew. Chem., Int. Ed. 1997, 36, 2108. doi:10.1002/anie.199721081 |
| 30. |
Goldfuss, B.; Kazmaier, U. Tetrahedron 2000, 56, 6493. doi:10.1016/S0040-4020(00)00613-X
The "trans phosphane preference" also explains memory effects in allylic substitutions. |
| 6. |
Pretot, R.; Pfaltz, A. Angew. Chem. 1998, 110, 337.
Angew. Chem., Int. Ed. 1998, 37, 323. doi:10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T |
| 26. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 5. | Sprinz, J.; Helmchen, G. Tetrahedron Lett. 1993, 34, 1769. doi:10.1016/S0040-4039(00)60774-8 |
| 26. | Kop-Weiershausen, T.; Lex, J.; Neudörfl, J.-M.; Goldfuss, B. Beilstein J. Org. Chem. 2005, 1, No. 6. doi:10.1186/1860-5397-1-6 |
| 3. | Delbecq, F.; Lapouge, C. Organometallics 2000, 19, 2716. doi:10.1021/om0003032 |
| 4. | Goldfuss, B. J. Organomet. Chem. 2006, 691, 4508–4513. doi:10.1016/j.jorganchem.2006.01.061 |
| 27. | Löschmann, T. Chirale P/N-Liganden: Fenchol-Derivate für enantioselektive Katalysen. Ph.D. Thesis, University of Heidelberg, Heidelberg, 2003. |
| 28. | Kop-Weiershausen, T. Biphenyl-Bisfenchol-Phosphan-Systeme (BIFOPs): Synthesen, strukturelle Studien & Einsatz in enantioselektiver Katalyse. Ph.D. Thesis, University of Cologne, Cologne, 2006. |
| 15. | Steigelmann, M.; Nisar, Y.; Rominger, F.; Goldfuss, B. Chem.–Eur. J. 2002, 8, 5211. doi:10.1002/1521-3765(20021115)8:22<5211::AID-CHEM5211>3.0.CO;2-S |
| 16. | Goldfuss, B.; Steigelmann, M.; Rominger, F. Eur. J. Org. Chem. 2000, 1785. doi:10.1002/(SICI)1099-0690(200005)2000:9<1785::AID-EJOC1785>3.0.CO;2-0 |
| 17. | Goldfuss, B.; Steigelmann, M. J. Mol. Model. 2000, 6, 166. doi:10.1007/s0089400060166 |
| 18. | Goldfuss, B.; Steigelmann, M.; Khan, S. I.; Houk, K. N. J. Org. Chem. 2000, 65, 77. doi:10.1021/jo991070v |
| 19. | Goldfuss, B.; Khan, S. I.; Houk, K. N. Organometallics 1999, 18, 2927. doi:10.1021/om990184u |
| 25. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273 |
| 10. | Lange, D.; Neudörfl, J.; Goldfuss, B. Tetrahedron 2006, 62, 3704. doi:10.1016/j.tet.2006.01.060 |
| 11. | Soki, F.; Neudörfl, J.-M.; Goldfuss, B. Tetrahedron 2005, 61, 10449. doi:10.1016/j.tet.2005.08.089 |
| 12. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2001, 7, 2028. doi:10.1002/1521-3765(20010504)7:9<2028::AID-CHEM2028>3.0.CO;2-Y |
| 13. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881. doi:10.1016/S0040-4020(99)01077-7 |
| 14. | Goldfuss, B.; Eisenträger, F. Aust. J. Chem. 2000, 53, 209. doi:10.1071/CH99184 |
| 13. | Goldfuss, B.; Rominger, F. Tetrahedron 2000, 56, 881. doi:10.1016/S0040-4020(99)01077-7 |
| 9. | Boele, M. D. K.; Kamer, P. C. J.; Lutz, M.; Spek, A. L.; de Vries, J. G.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Chem.–Eur. J. 2004, 10, 6232. doi:10.1002/chem.200400154 |
| 25. | Goldfuss, B.; Löschmann, T.; Rominger, F. Chem.–Eur. J. 2004, 10, 5422–5431. doi:10.1002/chem.200400273 |
| 8. | Hayashi, T. J. Organomet. Chem. 1999, 576, 195. doi:10.1016/S0022-328X(98)01058-4 |
| 20. | Goldfuss, B. Synthesis 2005, 2271. doi:10.1055/s-2005-872107 |
| 21. | Goldfuss, B.; Steigelmann, M.; Löschmann, T.; Schilling, G.; Rominger, F. Chem.–Eur. J. 2005, 11, 4019. doi:10.1002/chem.200500158 |
| 22. | Goldfuss, B. Enantioselective addition of organolithiums to C=O groups. In Organolithiums in Enantioselective Synthesis; Hodgson, D. M., Ed.; Topics in Organometallic Chemistry, Vol. 5; Springer: Berlin, 2003; pp 21–35. |
| 23. | Goldfuss, B.; Steigelmann, M.; Rominger, F.; Urtel, H. Chem.–Eur. J. 2001, 7, 4456. doi:10.1002/1521-3765(20011015)7:20<4456::AID-CHEM4456>3.0.CO;2-S |
| 24. |
Goldfuss, B.; Steigelmann, M.; Rominger, F. Angew. Chem. 2000, 112, 4299.
Angew. Chem., Int. Ed. 2000, 39, 4133. doi:10.1002/1521-3773(20001117)39:22<4133::AID-ANIE4133>3.0.CO;2-X |
© 2006 Goldfuss et al; licensee Beilstein-Institut
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)