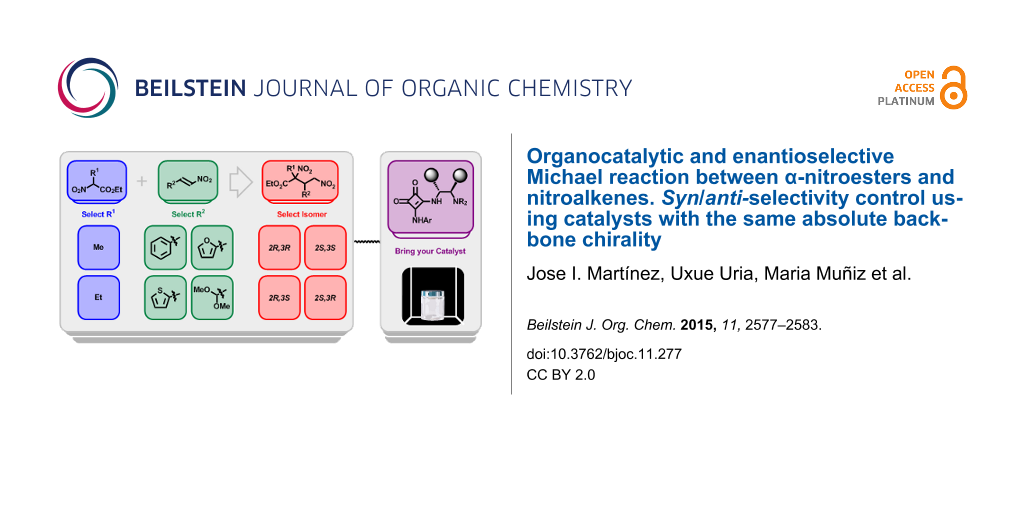
Abstract
The asymmetric and catalytic Michael reaction between α-nitroesters and nitroalkenes has been studied in the presence of two bifunctional catalysts both containing the same absolute chirality at the carbon backbone. The reaction performed in similar conditions allows us to control the syn or anti selectivity of the Michael adduct obtaining good yields and high enantiocontrol in all cases.

Graphical Abstract
Introduction
The absolute stereochemistry of a molecule has a paramount influence on the properties that this compound will have when interacting with biological systems [1]. As a consequence, the last decades have witnessed an enormous progress directed toward the development of synthetic methodology for the preparation of chiral molecules as single enantiomers with a well-defined three-dimensional arrangement. In this scenario, asymmetric catalysis arises as a key methodology for chemical production of enantiomerically enriched chiral compounds in terms of atom economy and reduced waste generation [2-6]. Nowadays, many very effective methodologies exist that allow the formation of a chiral compound as a single enantiomer. However, and despite the advances made in the field, an important challenge arises when a molecule containing multiple stereocentres has to be prepared because the access to all possible stereoisomers is not usually straightforward. While obtaining one or other mirror image of the target molecule can also be easily achieved by selecting the correct enantiomer of the catalyst, the relative configuration is typically governed by intrinsic factors associated to the mechanistic profile of the reaction and very often the formation of the major diastereoisomer is determined from the very beginning of the reaction and therefore access to any stereoisomer at will from the same set of starting materials with full absolute and relative stereocontrol is not trivial. Previous reports show that the diastereoselection can be directed by different approaches that include the modification of reaction conditions [7-9], the incorporation of additives or co-catalysts [10-13], the modification of some structural features of the substrate [14,15], the use of two catalysts that are structurally different to each other and that can also operate independently through a single transition state with minimal matched/mismatched interactions [16-22] or finally the concurrent use of two cycle-specific catalysts, in which each of them is exclusively involved in the formation of one stereocentre and therefore has to overcome the stereochemical bias exerted by the stereocentres generated in the previous steps [23-26].
In this context, we have recently reported a catalytic and enantioselective Michael/Michael cascade reaction in which α-nitro-δ-ketoesters react with nitroalkenes to provide densely functionalized cyclohexanes in excellent yield and stereoselectivity (Scheme 1) [27]. This reaction made use of bifunctional tertiary amine/squaramide catalysts [28-32] and, interestingly, we found that catalysts containing the same backbone chirality provided different diastereoisomers, thus allowing the development of a diastereodivergent process.
![[1860-5397-11-277-i1]](/bjoc/content/inline/1860-5397-11-277-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diastereodivergent cascade Michael/Michael reaction using catalysts with the same absolute chirality reported in our group.
Scheme 1: Diastereodivergent cascade Michael/Michael reaction using catalysts with the same absolute chiralit...
In this report, we wish to present the extension of this behaviour to the Michael reaction between α-substituted nitroacetates and nitroalkenes. The Michael reaction is regarded as a fundamental tool for the formation of C–C bonds during the synthesis of complex molecules [33-35], in particular, being able to control both the simple and the facial stereoselection of those versions in which two stereocentres are generated in the reaction become of special utility to synthetic chemists. In this case, using two different bifunctional tertiary amine/squaramide organocatalysts [36-61] with the same absolute chirality, the stereochemical outcome of the reaction can be controlled to provide the corresponding Michael adducts with two stereocentres, one of them being quaternary, and allowing the preparation of the desired diastereoisomer at will.
Results and Discussion
We started our work by first applying the conditions optimized for our recently reported cascade process to the reaction between ethyl 2-nitropropanoate (1a) and β-nitrostyrene (2a). As it can be seen in Scheme 2, the use of catalyst 4 led to the formation of the syn-diastereoisomer syn-3a in excellent yield, an acceptable 88:12 dr and 98% ee. As expected, the use of the pseudoenantiomeric catalyst 5 provided the corresponding enantiomer ent-syn-3a with similar results. Moving to cyclohexanediamine-based catalyst 6 resulted in the same behaviour as observed in our previous report, isolating the other possible diastereoisomer anti-3a in good yield, 88:12 dr and 90% ee.
![[1860-5397-11-277-i2]](/bjoc/content/inline/1860-5397-11-277-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Diastereodivergent enantioselective Michael reaction using ethyl 2-nitropropanoate and β-nitrostyrene as model substrates.
Scheme 2: Diastereodivergent enantioselective Michael reaction using ethyl 2-nitropropanoate and β-nitrostyre...
Once we had confirmed the good performance of the reaction in this intermolecular Michael reaction, we proceeded to evaluate the scope of this transformation in order to establish whether this method could be useful and general for a variety of nitroalkene Michael acceptors and nitroacetate donors. For this reason, we initially studied the reaction using squaramide 4 as catalyst that leads to the formation of syn-3 adducts. In this sense, and as it can be seen in Table 1, the reaction performed excellently in almost all the cases studied. In particular, when the reaction was conducted using nitrostyrene derivatives as Michael acceptors, the corresponding adducts 3a–k were isolated cleanly, in high yields, diastereo- and enantioselectivities regardless the electronic nature of the aromatic substituent of the nitrostyrene reagent. Indeed, the reaction using nitrostyrenes containing electron-donating groups at any of the position of the aryl ring led to the formation of the corresponding adducts (syn-3a–f) in excellent yield, good diastereoselectivities and enantiomeric excesses over 95% (Table 1, entries 2–6).
Table 1: Enantio- and diastereoselective Michael reaction between nitroesters 1 and nitroalkenes 2 catalysed by 4.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-277-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | Product | Yieldb | drc | ee (%)d |
|---|---|---|---|---|---|---|
| 1 | Me | Ph | syn-3a [27] | 92 | 88:12 | 98 |
| 2 | Me | 4-MeC6H4 | syn-3b | 85 | 89:11 | 97 |
| 3 | Me | 2-MeOC6H4 | syn-3c | 79 | 82:18 | 96 |
| 4 | Me | 3-MeOC6H4 | syn-3d | 92 | 88:12 | 96 |
| 5 | Me | 4-MeOC6H4 | syn-3e | 86 | 87:13 | >99 |
| 6 | Me | 4-BnOC6H4 | syn-3f | 85 | 86:14 | 97 |
| 7 | Me | 2-ClC6H4 | syn-3g | 80 | 88:12 | 98 |
| 8 | Me | 3-ClC6H4 | syn-3h | 92 | 86:14 | 97 |
| 9 | Me | 4-ClC6H4 | syn-3i | 97 | 88:12 | 97 |
| 10 | Me | 2-BrC6H4 | syn-3j | 75 | 85:15 | 97 |
| 11 | Me | 4-BrC6H4 | syn-3k | 92 | 87:13 | 97 |
| 12 | Me | 2-furyl | syn-3l | 80 | 86:14 | 93 |
| 13 | Me | 2-thienyl | syn-3m | 86 | 93:7 | 96 |
| 14 | Me | (MeO)2CH | syn-3n | 96 | 80:20 | 96 |
| 15 | Et | Ph | syn-3o | 92 | 86:14 | 98 |
| 16 | Et | 4-MeOC6H4 | syn-3p | 88 | 84:16 | 97 |
| 17 | Et | 4-BrC6H4 | syn-3q | 78 | 78:22 | 95 |
aAll reactions were carried out on a 0.1 mmol scale of 1 and 2 in toluene (1 M) at room temperature using 5.0 mol % of 4 as catalyst. bYield of pure Michael adducts as mixture of diastereoisomers after flash column chromatographic purification. cDetermined by 1H NMR analysis of the crude reaction mixture. dDetermined by HPLC on a chiral stationary phase (see experimental part in Supporting Information File 1 for details).
When nitrostyrenes containing electron-withdrawing groups were tested, the reaction proceeded equally well (Table 1, entries 7–11), although a slight decrease in the yield was observed for those cases in which the substituent was placed at the ortho-position (Table 1, entries 7 and 10). Heteroaromatic substituents at the nitroalkene reagent were also tested and those also reacted very efficiently, leading to the formation of adduct syn-3l–m in high yield and good stereoselection (Table 1, entries 12 and 13). Functionalized nitroalkene 2n also performed well in the reaction, providing the corresponding addition product syn-3n in good yield and high diastereo- and enantioselectivity (Table 1, entry 14). Disappointingly, when we tested β-alkyl-substituted nitroalkenes (R2 = iPr or n-Pr) we could not identify the formation of the desired addition product, and a complex mixture of products was obtained as a consequence of the very likely decomposition of these rather unstable nitroalkene reagents. Finally, we also surveyed the use of a nitroacetate donor with a bulkier substituent such as 1b which also performed very well in the reaction with trans-β-nitrostyrene (2a), para-methoxy-trans-β-nitrostyrene (2e) or para-bromo-trans-β-nitrostyrene (2k), yielding the corresponding products syn-3o–q with comparable results to those obtained with Michael donor 1a (Table 1, entries 15–17).
Next, we proceeded to evaluate the scope of the reaction leading to diastereomeric adducts anti-3 (Table 2) using bifunctional cyclohexanediamine/squaramide catalyst 6 which, as mentioned before, is based on a chiral backbone with the same absolute chirality as 4. The reaction conditions used for this reaction were those already observed to be optimal in our previous work for the cascade Michael/Henry reaction using this catalyst, that involved changing the solvent to 1,2-dichloroethane [27]. As it happened in the previous case, we could observe that the reaction performed well in all cases tested and, in general, with a similar level of chemical efficiency and stereocontrol, although in comparison with the 4-catalyzed version, enantioselectivities are normally around 5–10% lower if we analyse case by case. As it can be seen in Table 2, nitrostyrenes containing electron-withdrawing or electron-donating groups at the aromatic moiety performed similarly well in all cases, regardless the position of the substituents (Table 2, entries 1–11) and also heteroaryl moieties were well tolerated in the reaction (Table 2, entries 12 and 13). In the same line, functionalized nitroalkene 2n could also be successfully used in the reaction leading to adduct anti-3n in good yield, diastereo- and enantioselectivity (Table 2, entry 14). The use of bulkier ethyl 2-nitrobutanoate (1b) as Michael donor also led to good results for three representative nitroalkenes (Table 2, entries 15–17), although in the case of nitroalkene 2e a somewhat lower enantiomeric excess was obtained. Finally, and as it happened in the reaction catalyzed by 4, β-alkyl-substituted nitroalkenes were not suitable substrates for the reaction, probably due to their tendency to polymerize under the reaction conditions.
Table 2: Enantio- and diastereoselective Michael reaction between nitroesters 1 and nitroalkenes 2 catalysed by 6.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-277-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | Product | Yieldb | drc | ee (%)d |
|---|---|---|---|---|---|---|
| 1 | Me | Ph | anti-3a [27] | 98 | 88:12 | 90 |
| 2 | Me | 4-MeC6H4 | anti-3b [62] | 94 | 85:15 | 84 |
| 3 | Me | 2-MeOC6H4 | anti-3c | 95 | 90:10 | 86 |
| 4 | Me | 3-MeOC6H4 | anti-3d | 92 | 86:14 | 86 |
| 5 | Me | 4-MeOC6H4 | anti-3e [62] | 92 | 86:14 | 78 |
| 6 | Me | 4-BnOC6H4 | anti-3f | 90 | 86:14 | 81 |
| 7 | Me | 2-ClC6H4 | anti-3g [62] | 94 | 93:7 | 91 |
| 8 | Me | 3-ClC6H4 | anti-3h | 92 | 85:15 | 84 |
| 9 | Me | 4-ClC6H4 | anti-3i [62] | 91 | 83:17 | 84 |
| 10 | Me | 2-BrC6H4 | anti-3j [62] | 97 | 93:7 | 90 |
| 11 | Me | 4-BrC6H4 | anti-3k [62] | 93 | 83:17 | 84 |
| 12 | Me | 2-furyl | anti-3l [62] | 94 | 80:20 | 80 |
| 13 | Me | 2-thienyl | anti-3m | 98 | 87:13 | 80 |
| 14 | Me | (MeO)2CH | anti-3n | 96 | 76:24 | 84 |
| 15 | Et | Ph | anti-3o [62] | 94 | 87:13 | 90 |
| 16 | Et | 4-MeOC6H4 | anti-3p | 94 | 87:13 | 64 |
| 17 | Et | 4-BrC6H4 | anti-3q | 94 | 88:12 | 84 |
aAll reactions were carried out on a 0.1 mmol scale of 1 and 2 in 1,2-dichloroethane (1 M) at room temperature using 10.0 mol % of 6 as catalyst. bYield of pure Michael adducts as mixture of diastereoisomers after flash column chromatographic purification. cDetermined by 1H NMR analysis of the crude reaction mixture. dDetermined by HPLC on a chiral stationary phase (see experimental part in Supporting Information File 1 for details).
The absolute configuration was determined by X-ray analysis of products anti-3a and syn-3o for which suitable crystals could be obtained (Figure 1). The crystallographic analysis gave a (2R,3R) configuration for anti-3a [27] and a (2S,3R) for syn-3o [63] and this stereochemical assignment was extended to all other adducts prepared assuming an identical mechanistic pathway.
![[1860-5397-11-277-1]](/bjoc/content/figures/1860-5397-11-277-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP diagrams for anti-3a and syn-3o respectively.
Figure 1: ORTEP diagrams for anti-3a and syn-3o respectively.
This configuration is also in agreement with a transition state for the Michael reaction such as the one proposed in Scheme 3 [64]. This involves the activation of the nucleophile by the squaramide moiety of the bifunctional catalysts 4 and 6 through the formation of multiple H-bonding interactions and the simultaneous interaction between the nitroalkene acceptor and the ammonium salt moiety, the latter being generated after the initial deprotonation of the pronucleophile. The different possibilities offered by the two catalysts 4 and 6 to form geometrically different H-bonded complexes with the nitroacetate enolate would account for the different simple diastereoselection observed in each case, in which the nitronate moiety exposes a different reactive prochiral face. These results are in good agreement with our previously reported work in which DFT calculations also showed that the difference in the steric bulk of the nitrogen substituents of the Brønsted basic site of the catalysts (the quinuclidine moiety in catalyst 4 and the piperidine scaffold in catalyst 6) are the key parameters influencing this different arrangement for the nitroacetate pronucleophile [27].
![[1860-5397-11-277-i3]](/bjoc/content/inline/1860-5397-11-277-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed models to explain the stereochemical outcome of the reaction.
Scheme 3: Proposed models to explain the stereochemical outcome of the reaction.
Conclusion
We have developed an asymmetric catalytic diastereodivergent route for the synthesis of 2,4-dinitro esters taking advantage of the Michael addition of nitroalkenes and using two different bifunctional catalysts derived from cinchona alkaloids (catalyst 4) or cyclohexadiamine (catalyst 6). These catalysts, both with the same absolute backbone chirality, allow us to control the syn or anti selectivity obtaining the final products in good to excellent yields and with high enantioselectivity.
Supporting Information
| Supporting Information File 1: Experimental details, analytical data, NMR spectra and HPLC traces of all compounds prepared. | ||
| Format: PDF | Size: 7.6 MB | Download |
References
-
Brooks, W. H.; Guida, W. C.; Daniel, K. G. Curr. Top. Med. Chem. 2015, 11, 760. doi:10.2174/156802611795165098
Return to citation in text: [1] -
Ager, D. J.; de Vries, A. H. M.; de Vries, J. G. Chem. Soc. Rev. 2012, 41, 3340. doi:10.1039/c2cs15312b
Return to citation in text: [1] -
Howell, G. P. Org. Process Res. Dev. 2012, 16, 1258. doi:10.1021/op200381w
Return to citation in text: [1] -
Busacca, C. A.; Fandrick, D. R.; Song, J. J.; Senanayake, C. H. Adv. Synth. Catal. 2011, 353, 1825. doi:10.1002/adsc.201100488
Return to citation in text: [1] -
Blaser, H.-U.; Federsel, H.-J. Asymmetric Catalysis on Industrial Scale, 2nd ed.; Wiley-VCH: Weinheim, 2010. doi:10.1002/9783527630639
Return to citation in text: [1] -
Farina, V.; Reeves, J. T.; Senanayake, C. H.; Song, J. J. Chem. Rev. 2006, 106, 2734. doi:10.1021/cr040700c
Return to citation in text: [1] -
Enkisch, C.; Schneider, C. Eur. J. Org. Chem. 2009, 5549. doi:10.1002/ejoc.200900787
Return to citation in text: [1] -
Zhang, W.; Franzén, J. Adv. Synth. Catal. 2010, 352, 499. doi:10.1002/adsc.200900686
Return to citation in text: [1] -
Northrup, A. B.; MacMillan, D. W. C. Science 2004, 305, 1752. doi:10.1126/science.1101710
Return to citation in text: [1] -
Dou, X.; Lu, Y. Chem. – Eur. J. 2012, 18, 8315. doi:10.1002/chem.201200655
Return to citation in text: [1] -
Gao, J.; Bai, S.; Gao, Q.; Liu, Y.; Yang, Q. Chem. Commun. 2011, 47, 6716. doi:10.1039/c0cc05224h
Return to citation in text: [1] -
Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. J. Am. Chem. Soc. 2011, 133, 17934. doi:10.1021/ja207847p
Return to citation in text: [1] -
Bandini, M.; Cozzi, P. G.; Umani-Ronchi, A. Angew. Chem., Int. Ed. 2000, 39, 2327. doi:10.1002/1521-3773(20000703)39:13<2327::AID-ANIE2327>3.0.CO;2-9
Return to citation in text: [1] -
Marjanovic, J.; Divjakovic, V.; Matovic, R.; Ferjancic, Z.; Saicic, R. N. Eur. J. Org. Chem. 2013, 5555. doi:10.1002/ejoc.201300716
Return to citation in text: [1] -
Huber, J. D.; Leighton, J. L. J. Am. Chem. Soc. 2007, 129, 14552. doi:10.1021/ja076035h
Return to citation in text: [1] -
Oliveira, M. T.; Luparia, M.; Audisio, D.; Maulide, N. Angew. Chem., Int. Ed. 2013, 52, 13149. doi:10.1002/anie.201305933
Return to citation in text: [1] -
Audisio, D.; Luparia, M.; Oliveira, M. T.; Klütt, D.; Maulide, N. Angew. Chem., Int. Ed. 2012, 51, 7314. doi:10.1002/anie.201202853
Return to citation in text: [1] -
Lu, G.; Yoshino, T.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. Angew. Chem., Int. Ed. 2011, 50, 4382. doi:10.1002/anie.201101034
Return to citation in text: [1] -
Luparia, M.; Oliveira, M. T.; Audisio, D.; Frébault, F.; Goddard, R.; Maulide, N. Angew. Chem., Int. Ed. 2011, 50, 12631. doi:10.1002/anie.201106321
Return to citation in text: [1] -
Nojiri, A.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 3779. doi:10.1021/ja900084k
Return to citation in text: [1] -
Yan, X.-X.; Peng, Q.; Li, Q.; Zhang, K.; Yao, J.; Hou, X.-L.; Wu, Y.-D. J. Am. Chem. Soc. 2008, 130, 14362. doi:10.1021/ja804527r
Return to citation in text: [1] -
Wang, B.; Wu, F.; Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2007, 129, 768. doi:10.1021/ja0670409
Return to citation in text: [1] -
Rana, N. K.; Huang, H.; Zhao, J. C.-G. Angew. Chem., Int. Ed. 2014, 53, 7619. doi:10.1002/anie.201404072
Return to citation in text: [1] -
Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. doi:10.1126/science.1237068
Return to citation in text: [1] -
Simmons, B.; Walji, A. M.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2009, 48, 4349. doi:10.1002/anie.200900220
Return to citation in text: [1] -
Chi, Y.; Scroggins, S. T.; Fréchet, J. M. J. J. Am. Chem. Soc. 2008, 130, 6322. doi:10.1021/ja8013456
Return to citation in text: [1] -
Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416. doi:10.1021/ja805693p
See for a pioneering report on the use of bifunctional tertiary amine/squaramides as organocatalysts.
Return to citation in text: [1] -
Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Adv. Synth. Catal. 2015, 357, 253. doi:10.1002/adsc.201401003
Return to citation in text: [1] -
Tsakos, M.; Kokotos, C. G. Tetrahedron 2013, 69, 10199. doi:10.1016/j.tet.2013.09.080
Return to citation in text: [1] -
Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890. doi:10.1002/chem.201003694
Return to citation in text: [1] -
Türkmen, Y. E.; Zhu, Y.; Rawal, V. H. Brønsted Acids. In Comprehensive Enantioselective Organocatalysis; Dalko, P. I., Ed.; Wiley-VCH: Weinheim, 2013; Vol. 2, pp 241–288. doi:10.1002/9783527658862.ch10
Return to citation in text: [1] -
Vicario, J. L.; Badia, D.; Carrillo, L. Synthesis 2007, 2065. doi:10.1055/s-2007-983747
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Cui, L.-Y.; Wang, Y.-H.; Chen, S.-R.; Wang, Y.-M.; Zhou, Z.-H. RSC Adv. 2015, 5, 88133. doi:10.1039/C5RA17503H
Return to citation in text: [1] -
Wang, Z.-H.; Wu, Z.-J.; Huang, X.-Q.; Yue, D.-F.; You, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Chem. Commun. 2015, 51, 15835. doi:10.1039/C5CC06383C
Return to citation in text: [1] -
Zhang, Z.-P.; Dong, N.; Li, X.; Cheng, J.-P. Org. Biomol. Chem. 2015, 13, 9943. doi:10.1039/C5OB01691F
Return to citation in text: [1] -
Kim, H.; Kim, S.-G. Tetrahedron Lett. 2015, 56, 4819. doi:10.1016/j.tetlet.2015.06.071
Return to citation in text: [1] -
Bae, H. Y.; Song, C. E. ACS Catal. 2015, 5, 3613. doi:10.1021/acscatal.5b00685
Return to citation in text: [1] -
Chen, L.; You, Y.; Zhang, M.-L.; Zhao, J.-g.; Zuo, J.; Zhang, X.-M.; Yuan, W.-C.; Xu, X.-Y. Org. Biomol. Chem. 2015, 13, 4413. doi:10.1039/C5OB00317B
Return to citation in text: [1] -
Bera, K.; Namboothiri, I. N. N. J. Org. Chem. 2015, 80, 1402. doi:10.1021/jo502332r
Return to citation in text: [1] -
Işik, M.; Unver, M. Y.; Tanyeli, C. J. Org. Chem. 2015, 80, 828. doi:10.1021/jo5022597
Return to citation in text: [1] -
Rao, K. S.; Trivedi, R.; Kantam, M. L. Synlett 2015, 26, 221. doi:10.1055/s-0034-1379491
Return to citation in text: [1] -
Beswick, J.; Blanco, V.; De Bo, G.; Leigh, D. A.; Lewandowska, U.; Lewandowski, B.; Mishiro, K. Chem. Sci. 2015, 6, 140. doi:10.1039/C4SC03279A
Return to citation in text: [1] -
Zhong, Y.; Ma, S.; Xu, Z.; Chang, M.; Wang, R. RSC Adv. 2014, 4, 49930. doi:10.1039/C4RA09128K
Return to citation in text: [1] -
Liu, Y.; Wang, Q.; Wang, Y.; Song, H.; Zhou, Z. ChemCatChem 2014, 6, 2298. doi:10.1002/cctc.201402168
Return to citation in text: [1] -
Xu, X.; Cheng, T.; Liu, X.; Xu, J.; Jin, R.; Liu, G. ACS Catal. 2014, 4, 2137. doi:10.1021/cs5002459
Return to citation in text: [1] -
Pham, T. S.; Gönczi, K.; Kardos, G.; Süle, K.; Hegedűs, L.; Kállay, M.; Kubinyi, M.; Szabó, P.; Petneházy, I.; Tőke, L.; Jászai, Z. Tetrahedron: Asymmetry 2013, 24, 1605. doi:10.1016/j.tetasy.2013.10.008
Return to citation in text: [1] -
Zhao, M.-X.; Ji, F.-H.; Zhao, X.-L.; Han, Z.-Z.; Shi, M. Eur. J. Org. Chem. 2014, 644. doi:10.1002/ejoc.201301457
Return to citation in text: [1] -
Liu, B.; Han, X.; Dong, Z.; Lv, H.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2013, 24, 1276. doi:10.1016/j.tetasy.2013.08.010
Return to citation in text: [1] -
Baran, R.; Veverková, E.; Škvorcová, A.; Šebesta, R. Org. Biomol. Chem. 2013, 11, 7705. doi:10.1039/c3ob41709c
Return to citation in text: [1] -
Yang, K. S.; Nibbs, A. E.; Türkmen, Y. E.; Rawal, V. H. J. Am. Chem. Soc. 2013, 135, 16050. doi:10.1021/ja409012q
Return to citation in text: [1] -
Zhang, J.; Liu, X.; Ma, X.; Wang, R. Chem. Commun. 2013, 49, 9329. doi:10.1039/c3cc44059a
Return to citation in text: [1] -
Li, J.-H.; Du, D.-M. Org. Biomol. Chem. 2013, 11, 6215. doi:10.1039/c3ob41045e
Return to citation in text: [1] -
Kasaplar, P.; Rodriguez-Escrich, C.; Pericàs, M. A. Org. Lett. 2013, 15, 3498. doi:10.1021/ol400974z
Return to citation in text: [1] -
Mailhol, D.; del Mar Sanchez Duque, M.; Raimondi, W.; Bonne, D.; Constantieux, T.; Coquerel, Y.; Rodriguez, J. Adv. Synth. Catal. 2012, 354, 3523. doi:10.1002/adsc.201200658
Return to citation in text: [1] -
Kasaplar, P.; Riente, P.; Hartmann, C.; Pericàs, M. A. Adv. Synth. Catal. 2012, 354, 2905. doi:10.1002/adsc.201200526
Return to citation in text: [1] -
Noole, A.; Järving, I.; Werner, F.; Lopp, M.; Malkov, A.; Kanger, T. Org. Lett. 2012, 14, 4922. doi:10.1021/ol302245b
Return to citation in text: [1] -
Marcos, V.; Alemán, J.; Garcia Ruano, J. L.; Marini, F.; Tiecco, M. Org. Lett. 2011, 13, 3052. doi:10.1021/ol200923p
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450. doi:10.1021/ol102294g
Return to citation in text: [1] -
Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
CCDC 1416377 (syn-3o) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Kótai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Pápai, I.; Soós, T. Chem. – Eur. J. 2014, 20, 5631. doi:10.1002/chem.201304553
Return to citation in text: [1]
| 1. | Brooks, W. H.; Guida, W. C.; Daniel, K. G. Curr. Top. Med. Chem. 2015, 11, 760. doi:10.2174/156802611795165098 |
| 14. | Marjanovic, J.; Divjakovic, V.; Matovic, R.; Ferjancic, Z.; Saicic, R. N. Eur. J. Org. Chem. 2013, 5555. doi:10.1002/ejoc.201300716 |
| 15. | Huber, J. D.; Leighton, J. L. J. Am. Chem. Soc. 2007, 129, 14552. doi:10.1021/ja076035h |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 10. | Dou, X.; Lu, Y. Chem. – Eur. J. 2012, 18, 8315. doi:10.1002/chem.201200655 |
| 11. | Gao, J.; Bai, S.; Gao, Q.; Liu, Y.; Yang, Q. Chem. Commun. 2011, 47, 6716. doi:10.1039/c0cc05224h |
| 12. | Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. J. Am. Chem. Soc. 2011, 133, 17934. doi:10.1021/ja207847p |
| 13. | Bandini, M.; Cozzi, P. G.; Umani-Ronchi, A. Angew. Chem., Int. Ed. 2000, 39, 2327. doi:10.1002/1521-3773(20000703)39:13<2327::AID-ANIE2327>3.0.CO;2-9 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 7. | Enkisch, C.; Schneider, C. Eur. J. Org. Chem. 2009, 5549. doi:10.1002/ejoc.200900787 |
| 8. | Zhang, W.; Franzén, J. Adv. Synth. Catal. 2010, 352, 499. doi:10.1002/adsc.200900686 |
| 9. | Northrup, A. B.; MacMillan, D. W. C. Science 2004, 305, 1752. doi:10.1126/science.1101710 |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 2. | Ager, D. J.; de Vries, A. H. M.; de Vries, J. G. Chem. Soc. Rev. 2012, 41, 3340. doi:10.1039/c2cs15312b |
| 3. | Howell, G. P. Org. Process Res. Dev. 2012, 16, 1258. doi:10.1021/op200381w |
| 4. | Busacca, C. A.; Fandrick, D. R.; Song, J. J.; Senanayake, C. H. Adv. Synth. Catal. 2011, 353, 1825. doi:10.1002/adsc.201100488 |
| 5. | Blaser, H.-U.; Federsel, H.-J. Asymmetric Catalysis on Industrial Scale, 2nd ed.; Wiley-VCH: Weinheim, 2010. doi:10.1002/9783527630639 |
| 6. | Farina, V.; Reeves, J. T.; Senanayake, C. H.; Song, J. J. Chem. Rev. 2006, 106, 2734. doi:10.1021/cr040700c |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 28. |
Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416. doi:10.1021/ja805693p
See for a pioneering report on the use of bifunctional tertiary amine/squaramides as organocatalysts. |
| 29. | Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Adv. Synth. Catal. 2015, 357, 253. doi:10.1002/adsc.201401003 |
| 30. | Tsakos, M.; Kokotos, C. G. Tetrahedron 2013, 69, 10199. doi:10.1016/j.tet.2013.09.080 |
| 31. | Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890. doi:10.1002/chem.201003694 |
| 32. | Türkmen, Y. E.; Zhu, Y.; Rawal, V. H. Brønsted Acids. In Comprehensive Enantioselective Organocatalysis; Dalko, P. I., Ed.; Wiley-VCH: Weinheim, 2013; Vol. 2, pp 241–288. doi:10.1002/9783527658862.ch10 |
| 36. | Cui, L.-Y.; Wang, Y.-H.; Chen, S.-R.; Wang, Y.-M.; Zhou, Z.-H. RSC Adv. 2015, 5, 88133. doi:10.1039/C5RA17503H |
| 37. | Wang, Z.-H.; Wu, Z.-J.; Huang, X.-Q.; Yue, D.-F.; You, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Chem. Commun. 2015, 51, 15835. doi:10.1039/C5CC06383C |
| 38. | Zhang, Z.-P.; Dong, N.; Li, X.; Cheng, J.-P. Org. Biomol. Chem. 2015, 13, 9943. doi:10.1039/C5OB01691F |
| 39. | Kim, H.; Kim, S.-G. Tetrahedron Lett. 2015, 56, 4819. doi:10.1016/j.tetlet.2015.06.071 |
| 40. | Bae, H. Y.; Song, C. E. ACS Catal. 2015, 5, 3613. doi:10.1021/acscatal.5b00685 |
| 41. | Chen, L.; You, Y.; Zhang, M.-L.; Zhao, J.-g.; Zuo, J.; Zhang, X.-M.; Yuan, W.-C.; Xu, X.-Y. Org. Biomol. Chem. 2015, 13, 4413. doi:10.1039/C5OB00317B |
| 42. | Bera, K.; Namboothiri, I. N. N. J. Org. Chem. 2015, 80, 1402. doi:10.1021/jo502332r |
| 43. | Işik, M.; Unver, M. Y.; Tanyeli, C. J. Org. Chem. 2015, 80, 828. doi:10.1021/jo5022597 |
| 44. | Rao, K. S.; Trivedi, R.; Kantam, M. L. Synlett 2015, 26, 221. doi:10.1055/s-0034-1379491 |
| 45. | Beswick, J.; Blanco, V.; De Bo, G.; Leigh, D. A.; Lewandowska, U.; Lewandowski, B.; Mishiro, K. Chem. Sci. 2015, 6, 140. doi:10.1039/C4SC03279A |
| 46. | Zhong, Y.; Ma, S.; Xu, Z.; Chang, M.; Wang, R. RSC Adv. 2014, 4, 49930. doi:10.1039/C4RA09128K |
| 47. | Liu, Y.; Wang, Q.; Wang, Y.; Song, H.; Zhou, Z. ChemCatChem 2014, 6, 2298. doi:10.1002/cctc.201402168 |
| 48. | Xu, X.; Cheng, T.; Liu, X.; Xu, J.; Jin, R.; Liu, G. ACS Catal. 2014, 4, 2137. doi:10.1021/cs5002459 |
| 49. | Pham, T. S.; Gönczi, K.; Kardos, G.; Süle, K.; Hegedűs, L.; Kállay, M.; Kubinyi, M.; Szabó, P.; Petneházy, I.; Tőke, L.; Jászai, Z. Tetrahedron: Asymmetry 2013, 24, 1605. doi:10.1016/j.tetasy.2013.10.008 |
| 50. | Zhao, M.-X.; Ji, F.-H.; Zhao, X.-L.; Han, Z.-Z.; Shi, M. Eur. J. Org. Chem. 2014, 644. doi:10.1002/ejoc.201301457 |
| 51. | Liu, B.; Han, X.; Dong, Z.; Lv, H.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2013, 24, 1276. doi:10.1016/j.tetasy.2013.08.010 |
| 52. | Baran, R.; Veverková, E.; Škvorcová, A.; Šebesta, R. Org. Biomol. Chem. 2013, 11, 7705. doi:10.1039/c3ob41709c |
| 53. | Yang, K. S.; Nibbs, A. E.; Türkmen, Y. E.; Rawal, V. H. J. Am. Chem. Soc. 2013, 135, 16050. doi:10.1021/ja409012q |
| 54. | Zhang, J.; Liu, X.; Ma, X.; Wang, R. Chem. Commun. 2013, 49, 9329. doi:10.1039/c3cc44059a |
| 55. | Li, J.-H.; Du, D.-M. Org. Biomol. Chem. 2013, 11, 6215. doi:10.1039/c3ob41045e |
| 56. | Kasaplar, P.; Rodriguez-Escrich, C.; Pericàs, M. A. Org. Lett. 2013, 15, 3498. doi:10.1021/ol400974z |
| 57. | Mailhol, D.; del Mar Sanchez Duque, M.; Raimondi, W.; Bonne, D.; Constantieux, T.; Coquerel, Y.; Rodriguez, J. Adv. Synth. Catal. 2012, 354, 3523. doi:10.1002/adsc.201200658 |
| 58. | Kasaplar, P.; Riente, P.; Hartmann, C.; Pericàs, M. A. Adv. Synth. Catal. 2012, 354, 2905. doi:10.1002/adsc.201200526 |
| 59. | Noole, A.; Järving, I.; Werner, F.; Lopp, M.; Malkov, A.; Kanger, T. Org. Lett. 2012, 14, 4922. doi:10.1021/ol302245b |
| 60. | Marcos, V.; Alemán, J.; Garcia Ruano, J. L.; Marini, F.; Tiecco, M. Org. Lett. 2011, 13, 3052. doi:10.1021/ol200923p |
| 61. | Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450. doi:10.1021/ol102294g |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 23. | Rana, N. K.; Huang, H.; Zhao, J. C.-G. Angew. Chem., Int. Ed. 2014, 53, 7619. doi:10.1002/anie.201404072 |
| 24. | Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. doi:10.1126/science.1237068 |
| 25. | Simmons, B.; Walji, A. M.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2009, 48, 4349. doi:10.1002/anie.200900220 |
| 26. | Chi, Y.; Scroggins, S. T.; Fréchet, J. M. J. J. Am. Chem. Soc. 2008, 130, 6322. doi:10.1021/ja8013456 |
| 16. | Oliveira, M. T.; Luparia, M.; Audisio, D.; Maulide, N. Angew. Chem., Int. Ed. 2013, 52, 13149. doi:10.1002/anie.201305933 |
| 17. | Audisio, D.; Luparia, M.; Oliveira, M. T.; Klütt, D.; Maulide, N. Angew. Chem., Int. Ed. 2012, 51, 7314. doi:10.1002/anie.201202853 |
| 18. | Lu, G.; Yoshino, T.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. Angew. Chem., Int. Ed. 2011, 50, 4382. doi:10.1002/anie.201101034 |
| 19. | Luparia, M.; Oliveira, M. T.; Audisio, D.; Frébault, F.; Goddard, R.; Maulide, N. Angew. Chem., Int. Ed. 2011, 50, 12631. doi:10.1002/anie.201106321 |
| 20. | Nojiri, A.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 3779. doi:10.1021/ja900084k |
| 21. | Yan, X.-X.; Peng, Q.; Li, Q.; Zhang, K.; Yao, J.; Hou, X.-L.; Wu, Y.-D. J. Am. Chem. Soc. 2008, 130, 14362. doi:10.1021/ja804527r |
| 22. | Wang, B.; Wu, F.; Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2007, 129, 768. doi:10.1021/ja0670409 |
| 33. | Vicario, J. L.; Badia, D.; Carrillo, L. Synthesis 2007, 2065. doi:10.1055/s-2007-983747 |
| 34. | Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299. doi:10.1016/j.tetasy.2007.01.023 |
| 35. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701. doi:10.1002/ejoc.200600653 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 63. | CCDC 1416377 (syn-3o) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 64. | Kótai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Pápai, I.; Soós, T. Chem. – Eur. J. 2014, 20, 5631. doi:10.1002/chem.201304553 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 27. | Martínez, J. I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J. L. Adv. Synth. Catal. 2014, 356, 3627. doi:10.1002/adsc.201400502 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
| 62. | Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B. M.; Deng, L. Angew. Chem., Int. Ed. 2005, 44, 105. doi:10.1002/anie.200461923 |
© 2015 Martínez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)