Abstract
The donor–acceptor–π-conjugated (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2 with two (diphenylamino)carbazole thiophene units as D (electron-donating group)–π (π-conjugated bridge) moiety and a pyridine, pyrazine or triazine ring as electron-withdrawing group (electron-accepting group, A) have been designed and synthesized. The photophysical and electrochemical properties of the three dyes were investigated by photoabsorption and fluorescence spectroscopy, Lippert–Mataga plots, cyclic voltammetry and density functional theory calculations. The photoabsorption maximum (λmax,abs) and the fluorescence maximum (λmax,fl) for the intramolecular charge-transfer characteristic band of the (D–π–)2A fluorescent dyes show bathochromic shifts in the order of OUY-2 < OUK-2 < OUJ-2. Moreover, the photoabsorption bands of the (D–π–)2A fluorescent dyes are nearly independent of solvent polarity, while the fluorescence bands showed bathochromic shifts with increasing solvent polarity (i.e., positive fluorescence solvatochromism). The Lippert–Mataga plots for OUY-2, OUK-2 and OUJ-2 indicate that the Δμ (= μe − μg) value, which is the difference in the dipole moment of the dye between the excited (μe) and the ground (μg) states, increases in the order of OUY-2 < OUK-2 < OUJ-2. Therefore, the fact explains our findings that OUJ-2 shows large bathochromic shifts of the fluorescence maxima in polar solvents, as well as the Stokes shift values of OUJ-2 in polar solvents are much larger than those in nonpolar solvents. The cyclic voltammetry of OUY-2, OUK-2 and OUJ-2 demonstrated that there is little difference in the HOMO energy level among the three dyes, but the LUMO energy levels decrease in the order of OUY-2 > OUK-2 > OUJ-2. Consequently, this work reveals that for the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2 the bathochromic shifts of λmax,abs and λmax,fl and the lowering of the LUMO energy level are dependent on the electron-withdrawing ability of the azine ring, which increases in the order of OUY-2 < OUK-2 < OUJ-2.

Graphical Abstract
Introduction
Donor–π-conjugated–acceptor (D–π–A) dyes are constructed of an electron-donating group (D) such as a diphenyl or dialkylamino group and an electron-withdrawing group (electron-accepting group, A) such as a nitro, cyano, and carboxy group or an azine ring such as pyridine, pyrazine and triazine linked by π-conjugated bridges such as oligoenes and heterocycles. Thus, the D–π–A dyes exhibit intense photoabsorption and fluorescence emission properties based on the intramolecular charge transfer (ICT) excitation from the D moiety to the A moiety [1-4]. Moreover, the D–π–A structure possesses considerable structural characteristics: the increase in the electron-donating and electron-accepting abilities of the D and A moieties and the expansion of π conjugation, respectively, can lead to a decrease in the energy gap between the HOMO and LUMO because the highest occupied molecular orbital (HOMO) is localized over the π-conjugated system containing the D moiety, and the lowest unoccupied molecular orbital (LUMO) is localized over the A moiety. Thus, the photophysical and electrochemical properties based on the ICT characteristics of D–π–A dyes should be tuneable by not only the electron-donating ability of D and the electron-accepting ability of A, but also by the electronic characteristics of the π bridge. Consequently, the D–π–A dyes are of considerable practical concern as a useful fluorescence sensor for cation, anion and neural species [5-14], an efficient emitter for organic light emitting diodes (OLEDs) [15-24], and a promising photosensitizer for dye-sensitized solar cells (DSSCs) [25-34].
Thus, in this work, to gain insight into the photophysical and electrochemical properties of D–π–A fluorescent dyes with an azine ring as electron-withdrawing group, we have designed and synthesized the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2 with two (diphenylamino)carbazole thiophene units as the D–π moiety and a pyridine, pyrazine or triazine ring as the A moiety (Figure 1), although we have already reported the synthesis of (D–π–)2A fluorescent dyes OUY-2 [2] and OUK-2 [3,4] and their partial photopysical and electrochemical properties. One advantage of (D–π–)2A fluorescent dyes over other D–π–A fluorescent dyes is their broad and intense photoabsorption spectral features. Herein, based on photoabsorption and fluorescence spectroscopy, Lippert–Mataga plots, cyclic voltammetry and density functional theory (DFT) calculations, we reveal the photophysical and electrochemical properties of the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2.
![[1860-5397-15-167-1]](/bjoc/content/figures/1860-5397-15-167-1.png?scale=2.0&max-width=1024&background=FFFFFF)
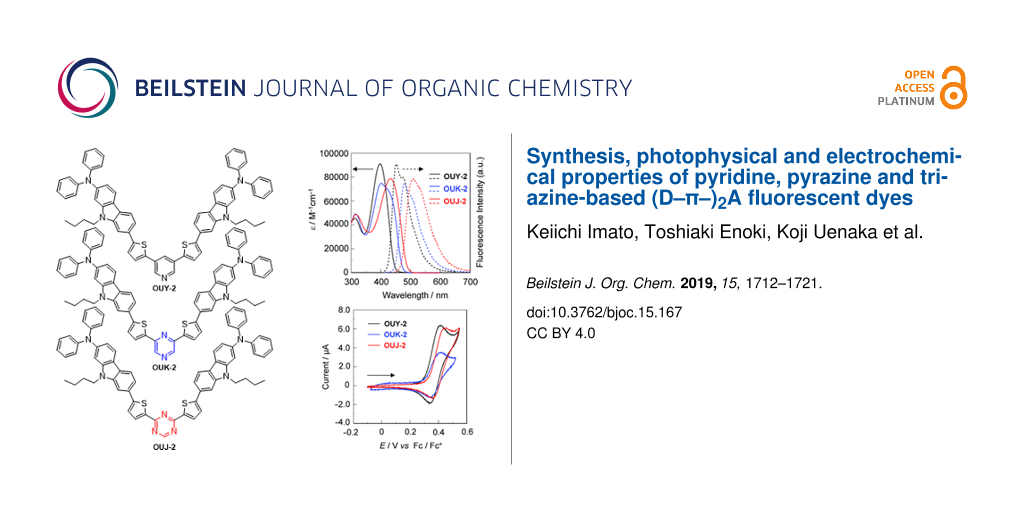
Figure 1: Chemical structures of the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2.
Figure 1: Chemical structures of the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2.
Results and Discussion
Synthesis
The (D–π–)2A fluorescent dyes OUY-2 [2], OUK-2 [3,4] and OUJ-2 were prepared by Stille coupling of stannyl compound 1 [3] with 3,5-dibromopyridine, 2,6-diiodopyrazine, and 2,4-dichloro-1,3,5-triazine, respectively (Scheme 1; see Experimental section for the synthetic procedure of OUJ-2).
![[1860-5397-15-167-i1]](/bjoc/content/inline/1860-5397-15-167-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of OUY-2, OUK-2 and OUJ-2.
Scheme 1: Synthesis of OUY-2, OUK-2 and OUJ-2.
Optical properties
The photoabsorption and fluorescence spectra of OUY-2, OUK-2 and OUJ-2 in various solvents are shown in Figure 2, and their optical data are summarized in Table 1. OUY-2, OUK-2 and OUJ-2 in toluene as a non-polar solvent show the photoabsorption maximum (λmax,abs) at 398 nm, 401 nm and 433 nm, respectively, which is assigned to the ICT excitation from the two (diphenylamino)carbazole thiophene units as D–π moiety to a pyridine, pyrazine or triazine ring as A moiety. For OUK-2, the shoulder band was observed at around 430 nm. Thus, the ICT-based photoabsorption band of the three dyes appears at a longer wavelength region in the order of OUY-2 < OUK-2 < OUJ-2, which is in agreement with the increase in the electron-withdrawing ability of the azine ring in the order of pyridyl group < pyrazyl group < triazyl group. The photoabsorption spectra of the three dyes are nearly independent of solvent polarity. This indicates that the electronic and structural characteristics of both the ground and Franck–Condon (FC) excited states do not differ much with a change in solvent polarity. The molar extinction coefficient (εmax) for the ICT band is ca. 100000 M−1 cm−1 for OUY-2, 75000 M−1 cm−1 for OUK-2 and 80000 M−1 cm−1 for OUJ-2. The corresponding fluorescence maximum (λmax,fl) of the three dyes in toluene also appears at a longer wavelength region in the order of OUY-2 (453 nm) < OUK-2 (480 nm) < OUJ-2 (509 nm). Interestingly, in contrast to the photoabsorption spectra, the fluorescence spectra are strongly dependent on the solvent polarity, that is, the three dyes showed a bathochromic shift of the fluorescence band with increasing solvent polarity from toluene to DMF (i.e., positive fluorescence solvatochromism). Thus, the Stokes shift (SS) values of the three dyes increase with increasing solvent polarity. Compared with OUY-2, OUK-2 and OUJ-2 exhibit significant fluorescence solvatochromic properties, that is, the two dyes show a significant decrease in the fluorescence quantum yield (Φf) in a polar solvent such as DMF (Φf = 0.59, 0.14 and 0.09 for OUY-2, OUK-2 and OUJ-2, respectively), although in relatively low polar solvents OUK-2 and OUJ-2 exhibit a higher Φf value (0.48–0.65 and 0.72–0.86, respectively) than OUY-2 (Φf = 0.38–0.58). For OUK-2 and OUJ-2, the large bathochromic shifts of the fluorescence band with a significant decrease in the Φf value in polar solvents such as DMF might be arising from the twisted intramolecular charge transfer (TICT) excited state due to the twisting between the pyrazyl or triazyl group and the (diphenylamino)carbazole thiophene moiety, leading to non-radiative deactivation [1]. On the other hand, it is worth mentioning here that the brightness values (ε × Φf) for OUY-2, OUK-2 and OUJ-2 in various solvents are fairly large (Table 1). Thus, the fact indicates that the (D–π–)2A fluorescent dyes have advantageous characteristics as emitters for OLEDs and fluorescence probes for biological imaging.
![[1860-5397-15-167-2]](/bjoc/content/figures/1860-5397-15-167-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Photoabsorption and (b) fluorescence (λex = ca. 400 nm) spectra of OUY-2 in various solvents. (c) Photoabsorption and (d) fluorescence (λex = ca. 400 nm) spectra of OUK-2 in various solvents. (e) Photoabsorption and (f) fluorescence (λex = ca. 430 nm) spectra of OUJ-2 in various solvents.
Figure 2: (a) Photoabsorption and (b) fluorescence (λex = ca. 400 nm) spectra of OUY-2 in various solvents. (...
Table 1: Optical data of OUY-2, OUK-2 and OUJ-2 in various solvents.
| Dye | Solvent | λmax,abs [nm] (ε [M−1cm−1]) | λmax,fl [nm] (Φf)a | Brightness [M−1cm−1] | Stokes shift [cm−1] |
|---|---|---|---|---|---|
| OUY-2 |
toluene
1,4-dioxane ethyl acetate THF DMF |
398 (91100)
398 (95800) 394 (98500) 397 (100000) 399 (97500) |
453 (0.38)
455 (0.40) 480 (0.39) 485 (0.58) 533 (0.59) |
34600
38300 38400 58000 57500 |
3050
3147 4547 4570 6300 |
| OUK-2 |
toluene
1,4-dioxane ethyl acetate THF DMF |
401 (74800)
397 (78300) 398 (75800) 394 (77400) 399 (73300) |
480 (0.48)
487 (0.62) 518 (0.55) 524 (0.65) 588 (0.14) |
35900
48500 41700 50300 10200 |
4104
4655 5820 6296 8055 |
| OUJ-2 |
toluene
1,4-dioxane ethyl acetate THF DMF |
433 (78500)
430 (85100) 428 (80100) 433 (81100) 435 (78900) |
509 (0.81)
525 (0.86) 568 (0.72) 576 (0.72) 665 (0.09) |
63600
73200 57700 58400 7100 |
3448
4208 5758 5733 7950 |
aFluorescence quantum yields (Φf) were determined by using a calibrated integrating sphere system (λex = 400 nm for OUY-2, 400 nm for OUK-2, and 430 nm for OUJ-2, respectively).
It is well accepted that the dipole–dipole interactions between the fluorescent dye and the solvent molecules are responsible for the solvent-dependent shifts in the fluorescence maxima [35-43]. Therefore, in order to understand the fluorescence solvatochromisms of OUY-2, OUK-2 and OUJ-2, we have investigated the relationships between the solvent polarity-dependent shift of the fluorescence maximum and the dipole moment of dye molecule on the basis of the Lippert–Mataga equation (Equation 1) [44-46]:
![[1860-5397-15-167-i2]](/bjoc/content/inline/1860-5397-15-167-i2.png?max-width=590&scale=1.18182)
where
![[1860-5397-15-167-i3]](/bjoc/content/inline/1860-5397-15-167-i3.png?max-width=590&scale=1.18182)
Consequently, on the basis of Equation (1) and Equation (2), the change in the dipole moment, Δμ = μe − μg, between the ground (μg) and the excited (μe) states can easily be evaluated from the slope of a plot of νst against Δf (the Lippert–Mataga plot), where νst is the Stokes shift (Table 1), ε0 is the vacuum permittivity, h is Planck’s constant, c is the velocity of light, a is the Onsager radius of the dye molecule (7.81 Å, 7.99 Å and 7.91 Å for OUY-2, OUK-2 and OUJ-2, respectively, estimated from DFT calculation at the B3LYP/6-31G(d,p) level of theory [47]), Δf is the orientation polarizability, ε is the static dielectric constant, and n is the refractive index of the solvent. The Lippert–Mataga plots (Figure 3) for the three dyes show high linearity, indicating that for the three dyes the solvent-dependent shift in the fluorescence maximum is mainly attributed to the dipole–dipole interactions between the dye molecule and the solvent molecule. The slopes (msl) became steep in the order of OUY-2 (10500 cm−1) < OUK-2 (12200 cm−1) < OUJ-2 (13700 cm−1). The correlation coefficient (R2) value for the calibration curve regarding the three dyes is 0.90 for OUY-2, 0.88 for OUK-2, and 0.89 for OUJ-2, which indicates good linearity. The Δμ values increase in the order of OUY-2 (22 D) < OUK-2 (25 D) < OUJ-2 (26 D), which corresponds to the increase in the electron-withdrawing ability of the azine rings (pyridyl group < pyrazyl group < triazyl group). Consequently, the Lippert–Mataga plots explains our findings that OUJ-2 shows large bathochromic shifts in its fluorescence maximum in polar solvents, as well as the SS values for OUJ-2 in polar solvents are much larger than those in nonpolar solvents (Table 1).
![[1860-5397-15-167-3]](/bjoc/content/figures/1860-5397-15-167-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Correlation of the Stokes shift (νst) and the orientation polarizability (Δf) according to Equation 1 and Equation 2, respectively, for OUY-2, OUK-2 and OUJ-2; solvent (ε, n, Δf): toluene (2.38, 1.4969, 0.0132), 1,4-dioxane (2.21, 1.4224, 0.0205), ethyl acetate (6.02, 1.3724, 0.199), THF (7.58, 1.4072, 0.2096) and DMF (36.71, 1.4305, 0.274) [4].
Figure 3: Correlation of the Stokes shift (νst) and the orientation polarizability (Δf) according to Equation 1 and Equation 2, r...
In order to investigate the solid-state photophysical properties of OUY-2, OUK-2 and OUJ-2, we have measured the solid-state fluorescence spectra of the solids (Figure 4). The λmax,fl of the as-recrystallized dyes appears at 550 nm for OUY-2, 592 nm for OUK-2, and 557 nm for OUJ-2, which showed a significant bathochromic shift by 97 nm, 112 nm, and 48 nm, respectively, compared with those in toluene. The solid-state Φf value is below 0.02 for OUY-2 and OUK-2 and 0.09 for OUJ-2, which are much lower than those in toluene. It is well known that D–π–A fluorescent dyes show bathochromic shifts of the λmax,fl and lower Φf values by changing from the solution state to the solid state. This fact is attributed to the delocalization of excitons or excimers due to the formation of intermolecular π–π interactions [48-51] between the dye molecules in the solid state, although we could not prepare single crystals of OUY-2, OUK-2 and OUJ-2 for the X-ray structural analysis.
![[1860-5397-15-167-4]](/bjoc/content/figures/1860-5397-15-167-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorescence spectra of OUY-2 (λex = 370 nm), OUK-2 (λex = 370 nm) and OUJ-2 (λex = 380 nm) in the solid state.
Figure 4: Fluorescence spectra of OUY-2 (λex = 370 nm), OUK-2 (λex = 370 nm) and OUJ-2 (λex = 380 nm) in the ...
Electrochemical properties
The electrochemical properties of OUY-2, OUK-2 and OUJ-2 were investigated by cyclic voltammetry (CV) in DMF containing 0.1 M tetrabutylammonium perchlorate (Bu4NClO4). The cyclic voltammograms of the three dyes are shown in Figure 5. The reversible oxidation waves (Epaox) for the three dyes were observed at 0.42 V for OUY-2 and OUK-2 and 0.45 V for OUJ-2, vs ferrocene/ferrocenium (Fc/Fc+) (Table 2). The corresponding reduction waves (Epcred) appeared at 0.35 V for OUY-2 and OUK-2 and 0.36 V for OUJ-2, thus indicating that the three dyes undergo an electrochemically stable oxidation–reduction process. The HOMO energy level (− [E1/2ox + 4.8] eV) versus the vacuum level was estimated from the half-wave potential for the oxidation (E1/2ox = 0.39 V for OUY-2 and OUK-2 and 0.40 V for OUJ-2). Therefore, the HOMO energy level was −5.19 eV for OUY-2 and OUK-2 and −5.20 eV for OUJ-2, respectively. This fact indicates that the three dyes have comparable HOMO energy levels. The LUMO energy level versus the vacuum level was evaluated from the E1/2ox and an intersection of photoabsorption and fluorescence spectra (449 nm; 2.76 eV for OUY-2, 481 nm; 2.58 eV for OUK-2, 506 nm; 2.45 eV for OUJ-2) in DMF. Consequently, the LUMO energy level was obtained through equation = [HOMO + E0–0] eV, where E0–0 transition energy is the intersection of the photoabsorption and fluorescence spectra corresponding to the optical energy gap between the HOMO and the LUMO. Thus, the LUMO energy level versus the vacuum level lowers in the order of OUY-2 (−2.43 eV) > OUK-2 (−2.61 eV) > OUJ-2 (−2.75 eV). This result demonstrates that an increase of the electron-withdrawing ability of the azine ring lowers the LUMO energy level of the (D–π–)2A fluorescent dyes. Consequently, the fact revealed that the bathochromic shift of the ICT-based photoabsorption band in the order of OUY-2 < OUK-2 < OUJ-2 is attributed to the stabilization of the LUMO energy level due to the increase in the electron-withdrawing ability of the azine ring in the order of pyridyl < pyrazyl < triazyl, resulting in a decrease in the energy gap between the HOMO and the LUMO.
![[1860-5397-15-167-5]](/bjoc/content/figures/1860-5397-15-167-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Cyclic voltammograms of OUY-2, OUK-2 and OUJ-2 in DMF containing 0.1 M Bu4NClO4. The arrow denotes the direction of the potential scan.
Figure 5: Cyclic voltammograms of OUY-2, OUK-2 and OUJ-2 in DMF containing 0.1 M Bu4NClO4. The arrow denotes ...
Table 2: Electrochemical data and HOMO and LUMO energy level of OUY-2, OUK-2 and OUJ-2.
| Dye | Epaox [V]a | Epcred [V]a | E1/2ox [V]a | HOMO [eV]b | LUMO [eV]c | E0–0 [eV]d |
|---|---|---|---|---|---|---|
| OUY-2 | 0.42 | 0.35 | 0.39 | −5.19 | −2.43 | 2.76 eV |
| OUK-2 | 0.42 | 0.35 | 0.39 | −5.19 | −2.61 | 2.58 eV |
| OUJ-2 | 0.45 | 0.36 | 0.40 | −5.20 | −2.75 | 2.45 eV |
aThe anodic peak (Epaox), the cathodic peak (Epcred) and the half-wave (E1/2ox) potentials for oxidation vs Fc/Fc+ were recorded in DMF/Bu4NClO4 (0.1 M) solution; bthe HOMO energy level (−[Eox1/2 + 4.8] eV) versus the vacuum level was evaluated from the E1/2ox for oxidation; cthe LUMO energy level versus the vacuum level was evaluated from the HOMO and the optical energy gap (E0–0), that is, the LUMO energy level was obtained through equation = [HOMO + E0–0] eV; dthe optical energy gap (E0–0) was determined from the intersection of the photoabsorption and fluorescence spectra in DMF.
Theoretical calculations
In order to examine the HOMO and LUMO distributions of OUY-2, OUK-2 and OUJ-2, the molecular structures and the molecular orbitals of the three dyes were calculated using the DFT at the B3LYP/6-31G(d,p) level of theory [47]. The results of the DFT calculation for the three dyes indicated that the HOMO is mostly localized on the two (diphenylamino)carbazole moieties containing the thiophene ring and the LUMO is mostly localized on the thienylpyridine moiety for OUY-2, the thienylpyrazine moiety for OUK-2 and the thienyltriazine moiety for OUJ-2 (Figure 6). Accordingly, the DFT calculations reveal that the photoexcitation of OUY-2, OUK-2 and OUJ-2 induces the ICT from the two (diphenylamino)carbazole moieties to each azine ring. The HOMO energy level of the three dyes is remarkably similar to each other (−4.80 eV, −4.78 eV and −4.84 eV for OUY-2, OUK-2 and OUJ-2, respectively), and the LUMO energy level is lowered in the order of OUY-2 (−1.56 eV) > OUK-2 (−1.76 eV) > OUJ-2 (−1.98 eV), which are in good agreement with the experimental results from the photoabsorption and fluorescence spectral analyses (Figure 2) and the cyclic voltammetry (Figure 5). Thus, the experimental results and the DFT calculation strongly demonstrated that the bathochromic shift of the ICT-based photoabsorption band in the order of OUY-2 < OUK-2 < OUJ-2 is attributed to a stabilization of the LUMO energy level due to the increase in the electron-withdrawing ability of the azine ring in the order of pyridyl < pyrazyl < triazyl.
![[1860-5397-15-167-6]](/bjoc/content/figures/1860-5397-15-167-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Energy level diagram, HOMO and LUMO of OUY-2, OUK-2 and OUJ-2, derived from the DFT calculations at the B3LYP/6-31G(d,p) level of theory. Numbers in parentheses are the experimental values.
Figure 6: Energy level diagram, HOMO and LUMO of OUY-2, OUK-2 and OUJ-2, derived from the DFT calculations at...
Conclusion
To gain insight into the photophysical and electrochemical properties of D–π–A fluorescent dyes with azine rings as electron-withdrawing groups, we have designed and synthesized a new type of (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2 with two (diphenylamino)carbazole thiophene units as the D (electron-donating group)–π (π-conjugated bridge) moiety and a pyridine, pyrazine or triazine ring as the electron-withdrawing group (electron-accepting group, A), and their photophysical and electrochemical properties were investigated. It was found that the intramolecular charge-transfer (ICT)-based photoabsorption and fluorescence bands of the three dyes appear at a longer wavelength region in the order of OUY-2 < OUK-2 < OUJ-2. This result is due to the increase in the electron-withdrawing ability of the azine ring in the order of pyridyl < pyrazyl < triazyl. Moreover, the (D–π–)2A fluorescent dyes showed a large bathochromic shift of the fluorescence maxima with increasing solvent polarity (i.e., positive fluorescence solvatochromism). The Lippert–Mataga plots revealed that the difference in the dipole moment of the dye between the excited state and the ground state increases in the order of OUY-2 < OUK-2 < OUJ-2. Thus, the fact explains our findings that OUJ-2 shows large bathochromic shifts of fluorescence maxima in polar solvents, as well as the Stokes shift values for OUJ-2 in polar solvents are much larger than those in nonpolar solvents. Cyclic voltammetry and DFT calculations demonstrated that the HOMO energy levels of the three dyes are remarkably similar, but the LUMO energy level is lowered in the order of OUY-2 > OUK-2 > OUJ-2, showing that increasing the electron-withdrawing ability of the azine ring lowers the LUMO energy level of the (D–π–)2A fluorescent dyes. Consequently, this work reveals that for the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2, the bathochromic shift of photoabsorption and fluorescence maxima and the lowering of the LUMO energy levels are dependent on the electron-withdrawing ability of the azine ring which increases in the order of OUY-2 < OUK-2 < OUJ-2.
Experimental
General methods
Melting points were measured with a Yanaco micro melting point apparatus MP model. FTIR spectra were recorded on a Shimadzu IRAffinity-1 spectrometer by ATR method. High-resolution mass spectra were acquired on a Thermo Fisher Scientific LTQ Orbitrap XL. 1H NMR and 13C NMR spectra were recorded on a Varian-400 (400 MHz) FT NMR spectrometer. Photoabsorption spectra were measured with a Hitachi U-2910 spectrophotometer, and fluorescence spectra were measured with a Horiba FluoroMax-4 spectrofluorometer. The fluorescence quantum yields in solution were determined by a Horiba FluoroMax-4 spectrofluorometer by using a calibrated integrating sphere system. Cyclic voltammetry (CV) curves were recorded in DMF/Bu4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ as reference electrode, a Pt plate as working electrode, and Pt wire as counter electrode by using an electrochemical measurement system HZ-7000 (Hokuto Denko).
Synthesis
General synthetic procedure of (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2
OUY-2 [2], OUK-2 [3] and OUJ-2 were prepared by Stille coupling of stannyl compound 1 [3] with 3,5-dibromopyridine, 2,6-diiodopyrazine, and 2,4-dichloro-1,3,5-triazine, respectively, by using Pd(PPh3)4 as a catalyst in toluene at 110 °C under an argon atmosphere (Scheme 1).
Synthesis of OYJ-2: A solution of 1 [3] (0.60 g, 0.95 mmol), 2,4-dichloro-1,3,5-triazine (0.071 g, 0.48 mmol), and Pd(PPh3)4 (0.18 g, 0.16 mmol) in toluene (10 mL) was stirred for 48 h at 110 °C under an argon atmosphere. After concentrating under reduced pressure, the resulting residue was dissolved in dichloromethane and washed with water. The dichloromethane extract was evaporated under reduced pressure. The residue was chromatographed on silica gel (ethyl acetate/dichloromethane 1:4 as eluent) to give OUJ-2 (0.38 g, yield 70%) as yellow solid; mp 267–269 °C; IR (ATR) ν̃: 1594, 1548, 1491 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 0.89 (t, J = 7.3 Hz, 6H), 1.29–1.35 (m, 4H), 1.75–1.83 (m, 4H), 4.22 (t, J = 7.1 Hz, 4H), 6.96 (dd, J = 1.8 and 8.4 Hz, 2H), 7.02–7.06 (m, 4H), 7.13–7.16 (m, 10H), 7.26–7.30 (m, 8H), 7.57 (d, J = 4.0 Hz, 2H), 7.60–7.63 (dd, J = 8.1 and 1.5 Hz, 2H), 7.72 (s, 2H), 7.95 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 8.1 Hz, 2H), 8.27 (d, J = 4.0 Hz, 2H), 9.00 (s, 1H) ppm; 13C NMR (100 MHz, CD2Cl2) δ 14.04, 20.86, 31.47, 43.10, 105.17, 106.55, 117.52, 117.92, 118.47, 120.53, 121.40, 123.08, 123.94, 124.50, 124.79, 129.58, 130.62, 133.57, 139.41, 141.56, 142.95, 147.31, 148.55, 153.55, 167.62 ppm (one aromatic carbon signal was not observed due to overlapping resonances); HRMS–ESI (m/z): [M + H] + calcd. for C67H56N7S2, 1022.40331; found, 1022.40344.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra of OUJ-2. | ||
| Format: PDF | Size: 221.3 KB | Download |
References
-
Ooyama, Y.; Uenaka, K.; Ohshita, J. RSC Adv. 2015, 5, 21012–21018. doi:10.1039/c4ra16399k
Return to citation in text: [1] [2] -
Ooyama, Y.; Uenaka, K.; Ohshita, J. Eur. J. Org. Chem. 2015, 3713–3720. doi:10.1002/ejoc.201500341
Return to citation in text: [1] [2] [3] [4] -
Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Enoki, T.; Ohshita, J.; Ooyama, Y. Bull. Chem. Soc. Jpn. 2018, 91, 1704–1709. doi:10.1246/bcsj.20180210
Return to citation in text: [1] [2] [3] [4] -
Guliyev, R.; Coskun, A.; Akkaya, E. U. J. Am. Chem. Soc. 2009, 131, 9007–9013. doi:10.1021/ja902584a
Return to citation in text: [1] -
Woodford, C. R.; Frady, E. P.; Smith, R. S.; Morey, B.; Canzi, G.; Palida, S. F.; Araneda, R. C.; Kristan, W. B., Jr.; Kubiak, C. P.; Miller, E. W.; Tsien, R. Y. J. Am. Chem. Soc. 2015, 137, 1817–1824. doi:10.1021/ja510602z
Return to citation in text: [1] -
Escudero, D. Acc. Chem. Res. 2016, 49, 1816–1824. doi:10.1021/acs.accounts.6b00299
Return to citation in text: [1] -
Saha, M. L.; Yan, X.; Stang, P. J. Acc. Chem. Res. 2016, 49, 2527–2539. doi:10.1021/acs.accounts.6b00416
Return to citation in text: [1] -
Mahendran, V.; Pasumpon, K.; Thimmarayaperumal, S.; Thilagar, P.; Shanmugam, S. J. Org. Chem. 2016, 81, 3597–3602. doi:10.1021/acs.joc.6b00267
Return to citation in text: [1] -
Sandeep, A.; Praveen, V. K.; Kartha, K. K.; Karunakaran, V.; Ajayaghosh, A. Chem. Sci. 2016, 7, 4460–4467. doi:10.1039/c6sc00629a
Return to citation in text: [1] -
Black, H. T.; Pelse, I.; Wolfe, R. M. W.; Reynolds, J. R. Chem. Commun. 2016, 52, 12877–12880. doi:10.1039/c6cc06443d
Return to citation in text: [1] -
Ji, L.; Griesbeck, S.; Marder, T. B. Chem. Sci. 2017, 8, 846–863. doi:10.1039/c6sc04245g
Return to citation in text: [1] -
Xu, Y.; Yu, S.; Chen, Q.; Chen, X.; Li, Y.; Yu, X.; Pu, L. Chem. – Eur. J. 2016, 22, 12061–12067. doi:10.1002/chem.201601540
Return to citation in text: [1] -
Zhou, J.; Outlaw, V. K.; Townsend, C. A.; Bragg, A. E. Chem. – Eur. J. 2016, 22, 15212–15215. doi:10.1002/chem.201603284
Return to citation in text: [1] -
Lin, S.-L.; Chan, L.-H.; Lee, R.-H.; Yen, M.-Y.; Kuo, W.-J.; Chen, C.-T.; Jeng, R.-J. Adv. Mater. (Weinheim, Ger.) 2008, 20, 3947–3952. doi:10.1002/adma.200801023
Return to citation in text: [1] -
Park, I. S.; Komiyama, H.; Yasuda, T. Chem. Sci. 2017, 8, 953–960. doi:10.1039/c6sc03793c
Return to citation in text: [1] -
Duan, C.; Li, J.; Han, C.; Ding, D.; Yang, H.; Wei, Y.; Xu, H. Chem. Mater. 2016, 28, 5667–5679. doi:10.1021/acs.chemmater.6b01691
Return to citation in text: [1] -
Huang, J.-J.; Hung, Y.-H.; Ting, P.-L.; Tsai, Y.-N.; Gao, H.-J.; Chiu, T.-L.; Lee, J.-H.; Chen, C.-L.; Chou, P.-T.; Leung, M.-k. Org. Lett. 2016, 18, 672–675. doi:10.1021/acs.orglett.5b03631
Return to citation in text: [1] -
Zhang, Q.; Li, J.; Shizu, K.; Huang, S.; Hirata, S.; Miyazaki, H.; Adachi, C. J. Am. Chem. Soc. 2012, 134, 14706–14709. doi:10.1021/ja306538w
Return to citation in text: [1] -
Feuillastre, S.; Pauton, M.; Gao, L.; Desmarchelier, A.; Riives, A. J.; Prim, D.; Tondelier, D.; Geffroy, B.; Muller, G.; Clavier, G.; Pieters, G. J. Am. Chem. Soc. 2016, 138, 3990–3993. doi:10.1021/jacs.6b00850
Return to citation in text: [1] -
Hirai, H.; Nakajima, K.; Nakatsuka, S.; Shiren, K.; Ni, J.; Nomura, S.; Ikuta, T.; Hatakeyama, T. Angew. Chem., Int. Ed. 2015, 54, 13581–13585. doi:10.1002/anie.201506335
Return to citation in text: [1] -
Sasabe, H.; Hayasaka, Y.; Komatsu, R.; Nakao, K.; Kido, J. Chem. – Eur. J. 2017, 23, 114–119. doi:10.1002/chem.201604303
Return to citation in text: [1] -
Yao, L.; Zhang, S.; Wang, R.; Li, W.; Shen, F.; Yang, B.; Ma, Y. Angew. Chem., Int. Ed. 2014, 53, 2119–2123. doi:10.1002/anie.201308486
Return to citation in text: [1] -
Suzuki, K.; Kubo, S.; Shizu, K.; Fukushima, T.; Wakamiya, A.; Murata, Y.; Adachi, C.; Kaji, H. Angew. Chem., Int. Ed. 2015, 54, 15231–15235. doi:10.1002/anie.201508270
Return to citation in text: [1] -
Mishra, A.; Fischer, M. K. R.; Bäuerle, P. Angew. Chem., Int. Ed. 2009, 48, 2474–2499. doi:10.1002/anie.200804709
Return to citation in text: [1] -
Ooyama, Y.; Harima, Y. Eur. J. Org. Chem. 2009, 2903–2934. doi:10.1002/ejoc.200900236
Return to citation in text: [1] -
Li, X.; Zheng, Z.; Jiang, W.; Wu, W.; Wang, Z.; Tian, H. Chem. Commun. 2015, 51, 3590–3592. doi:10.1039/c4cc08539f
Return to citation in text: [1] -
Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Kyomen, T.; Hanaya, M. Chem. Commun. 2015, 51, 6315–6317. doi:10.1039/c5cc00464k
Return to citation in text: [1] -
Wu, J.; Li, G.; Zhang, L.; Zhou, G.; Wang, Z.-S. J. Mater. Chem. A 2016, 4, 3342–3355. doi:10.1039/c5ta09763k
Return to citation in text: [1] -
Gao, Y.; Li, X.; Hu, Y.; Fan, Y.; Yuan, J.; Robertson, N.; Hua, J.; Marder, S. R. J. Mater. Chem. A 2016, 4, 12865–12877. doi:10.1039/c6ta05588e
Return to citation in text: [1] -
Yao, Z.; Zhang, M.; Li, R.; Yang, L.; Qiao, Y.; Wang, P. Angew. Chem., Int. Ed. 2015, 54, 5994–5998. doi:10.1002/anie.201501195
Return to citation in text: [1] -
Brogdon, P.; Giordano, F.; Puneky, G. A.; Dass, A.; Zakeeruddin, S. M.; Nazeeruddin, M. K.; Grätzel, M.; Tschumper, G. S.; Delcamp, J. H. Chem. – Eur. J. 2016, 22, 694–703. doi:10.1002/chem.201503187
Return to citation in text: [1] -
Ooyama, Y.; Inoue, S.; Nagano, T.; Kushimoto, K.; Ohshita, J.; Imae, I.; Komaguchi, K.; Harima, Y. Angew. Chem., Int. Ed. 2011, 50, 7429–7433. doi:10.1002/anie.201102552
Return to citation in text: [1] -
Ooyama, Y.; Sato, T.; Harima, Y.; Ohshita, J. J. Mater. Chem. A 2014, 2, 3293–3296. doi:10.1039/c3ta15067d
Return to citation in text: [1] -
Sumalekshmy, S.; Gopidas, K. R. J. Phys. Chem. B 2004, 108, 3705–3712. doi:10.1021/jp022549l
Return to citation in text: [1] -
Sumalekshmy, S.; Gopidas, K. R. New J. Chem. 2005, 29, 325–331. doi:10.1039/b409411e
Return to citation in text: [1] -
Sumalekshmy, S.; Gopidas, K. R. Photochem. Photobiol. Sci. 2005, 4, 539–546. doi:10.1039/b503251b
Return to citation in text: [1] -
Dias, F. B.; Pollock, S.; Hedley, G.; Pålsson, L.-O.; Monkman, A.; Perepichka, I. I.; Perepichka, I. F.; Tavasli, M.; Bryce, M. R. J. Phys. Chem. B 2006, 110, 19329–19339. doi:10.1021/jp0643653
Return to citation in text: [1] -
Zhao, G.-J.; Chen, R.-K.; Sun, M.-T.; Liu, J.-Y.; Li, G.-Y.; Gao, Y.-L.; Han, K.-L.; Yang, X.-C.; Sun, L. Chem. – Eur. J. 2008, 14, 6935–6947. doi:10.1002/chem.200701868
Return to citation in text: [1] -
Aronica, C.; Venancio-Marques, A.; Chauvin, J.; Robert, V.; Lemercier, G. Chem. – Eur. J. 2009, 15, 5047–5055. doi:10.1002/chem.200802325
Return to citation in text: [1] -
Butler, R. S.; Cohn, P.; Tenzel, P.; Abboud, K. A.; Castellano, R. K. J. Am. Chem. Soc. 2009, 131, 623–633. doi:10.1021/ja806348z
Return to citation in text: [1] -
Ooyama, Y.; Ito, G.; Kushimoto, K.; Komaguchi, K.; Imae, I.; Harima, Y. Org. Biomol. Chem. 2010, 8, 2756–2770. doi:10.1039/c003526b
Return to citation in text: [1] -
Enoki, T.; Matsuo, K.; Ohshita, J.; Ooyama, Y. Phys. Chem. Chem. Phys. 2017, 19, 3565–3574. doi:10.1039/c6cp08573c
Return to citation in text: [1] -
Lippert, E. Z. Naturforsch., A: Astrophys., Phys. Phys. Chem. 1955, 10, 541–545. doi:10.1515/zna-1955-0707
Return to citation in text: [1] -
Mataga, N.; Kaifu, Y.; Koizumi, M. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. doi:10.1246/bcsj.29.465
Return to citation in text: [1] -
Makitra, R. G. Reichardt, C., Solvents and Solvent Effects in Organic Chemistry, Weinheim: Wiley-VCH, 2003, 630 p.; Russian Journal of General Chemistry, Vol. 75; Springer Science and Business Media LLC, 2005; pp 664 ff. doi:10.1007/s11176-005-0294-y
Return to citation in text: [1] -
Both the geometry optimization and energy calculation were performed by employing density functional theory (DFT), at the level of B3LYP/6-31G(d,p) on the Gaussian09 program package, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009.
Return to citation in text: [1] [2] -
Langhals, H.; Potrawa, T.; Nöth, H.; Linti, G. Angew. Chem., Int. Ed. Engl. 1989, 28, 478–480. doi:10.1002/anie.198904781
Return to citation in text: [1] -
Yeh, H.-C.; Wu, W.-C.; Wen, Y.-S.; Dai, D.-C.; Wang, J.-K.; Chen, C.-T. J. Org. Chem. 2004, 69, 6455–6462. doi:10.1021/jo049512c
Return to citation in text: [1] -
Ooyama, Y.; Okamoto, T.; Yamaguchi, T.; Suzuki, T.; Hayashi, A.; Yoshida, K. Chem. – Eur. J. 2006, 12, 7827–7838. doi:10.1002/chem.200600094
Return to citation in text: [1] -
Ooyama, Y.; Hagiwara, Y.; Oda, Y.; Fukuoka, H.; Ohshita, J. RSC Adv. 2014, 4, 1163–1167. doi:10.1039/c3ra45785k
Return to citation in text: [1]
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 2. | Ooyama, Y.; Uenaka, K.; Ohshita, J. Eur. J. Org. Chem. 2015, 3713–3720. doi:10.1002/ejoc.201500341 |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 1. | Ooyama, Y.; Uenaka, K.; Ohshita, J. RSC Adv. 2015, 5, 21012–21018. doi:10.1039/c4ra16399k |
| 2. | Ooyama, Y.; Uenaka, K.; Ohshita, J. Eur. J. Org. Chem. 2015, 3713–3720. doi:10.1002/ejoc.201500341 |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 4. | Enoki, T.; Ohshita, J.; Ooyama, Y. Bull. Chem. Soc. Jpn. 2018, 91, 1704–1709. doi:10.1246/bcsj.20180210 |
| 2. | Ooyama, Y.; Uenaka, K.; Ohshita, J. Eur. J. Org. Chem. 2015, 3713–3720. doi:10.1002/ejoc.201500341 |
| 48. | Langhals, H.; Potrawa, T.; Nöth, H.; Linti, G. Angew. Chem., Int. Ed. Engl. 1989, 28, 478–480. doi:10.1002/anie.198904781 |
| 49. | Yeh, H.-C.; Wu, W.-C.; Wen, Y.-S.; Dai, D.-C.; Wang, J.-K.; Chen, C.-T. J. Org. Chem. 2004, 69, 6455–6462. doi:10.1021/jo049512c |
| 50. | Ooyama, Y.; Okamoto, T.; Yamaguchi, T.; Suzuki, T.; Hayashi, A.; Yoshida, K. Chem. – Eur. J. 2006, 12, 7827–7838. doi:10.1002/chem.200600094 |
| 51. | Ooyama, Y.; Hagiwara, Y.; Oda, Y.; Fukuoka, H.; Ohshita, J. RSC Adv. 2014, 4, 1163–1167. doi:10.1039/c3ra45785k |
| 25. | Mishra, A.; Fischer, M. K. R.; Bäuerle, P. Angew. Chem., Int. Ed. 2009, 48, 2474–2499. doi:10.1002/anie.200804709 |
| 26. | Ooyama, Y.; Harima, Y. Eur. J. Org. Chem. 2009, 2903–2934. doi:10.1002/ejoc.200900236 |
| 27. | Li, X.; Zheng, Z.; Jiang, W.; Wu, W.; Wang, Z.; Tian, H. Chem. Commun. 2015, 51, 3590–3592. doi:10.1039/c4cc08539f |
| 28. | Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Kyomen, T.; Hanaya, M. Chem. Commun. 2015, 51, 6315–6317. doi:10.1039/c5cc00464k |
| 29. | Wu, J.; Li, G.; Zhang, L.; Zhou, G.; Wang, Z.-S. J. Mater. Chem. A 2016, 4, 3342–3355. doi:10.1039/c5ta09763k |
| 30. | Gao, Y.; Li, X.; Hu, Y.; Fan, Y.; Yuan, J.; Robertson, N.; Hua, J.; Marder, S. R. J. Mater. Chem. A 2016, 4, 12865–12877. doi:10.1039/c6ta05588e |
| 31. | Yao, Z.; Zhang, M.; Li, R.; Yang, L.; Qiao, Y.; Wang, P. Angew. Chem., Int. Ed. 2015, 54, 5994–5998. doi:10.1002/anie.201501195 |
| 32. | Brogdon, P.; Giordano, F.; Puneky, G. A.; Dass, A.; Zakeeruddin, S. M.; Nazeeruddin, M. K.; Grätzel, M.; Tschumper, G. S.; Delcamp, J. H. Chem. – Eur. J. 2016, 22, 694–703. doi:10.1002/chem.201503187 |
| 33. | Ooyama, Y.; Inoue, S.; Nagano, T.; Kushimoto, K.; Ohshita, J.; Imae, I.; Komaguchi, K.; Harima, Y. Angew. Chem., Int. Ed. 2011, 50, 7429–7433. doi:10.1002/anie.201102552 |
| 34. | Ooyama, Y.; Sato, T.; Harima, Y.; Ohshita, J. J. Mater. Chem. A 2014, 2, 3293–3296. doi:10.1039/c3ta15067d |
| 47. | Both the geometry optimization and energy calculation were performed by employing density functional theory (DFT), at the level of B3LYP/6-31G(d,p) on the Gaussian09 program package, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009. |
| 15. | Lin, S.-L.; Chan, L.-H.; Lee, R.-H.; Yen, M.-Y.; Kuo, W.-J.; Chen, C.-T.; Jeng, R.-J. Adv. Mater. (Weinheim, Ger.) 2008, 20, 3947–3952. doi:10.1002/adma.200801023 |
| 16. | Park, I. S.; Komiyama, H.; Yasuda, T. Chem. Sci. 2017, 8, 953–960. doi:10.1039/c6sc03793c |
| 17. | Duan, C.; Li, J.; Han, C.; Ding, D.; Yang, H.; Wei, Y.; Xu, H. Chem. Mater. 2016, 28, 5667–5679. doi:10.1021/acs.chemmater.6b01691 |
| 18. | Huang, J.-J.; Hung, Y.-H.; Ting, P.-L.; Tsai, Y.-N.; Gao, H.-J.; Chiu, T.-L.; Lee, J.-H.; Chen, C.-L.; Chou, P.-T.; Leung, M.-k. Org. Lett. 2016, 18, 672–675. doi:10.1021/acs.orglett.5b03631 |
| 19. | Zhang, Q.; Li, J.; Shizu, K.; Huang, S.; Hirata, S.; Miyazaki, H.; Adachi, C. J. Am. Chem. Soc. 2012, 134, 14706–14709. doi:10.1021/ja306538w |
| 20. | Feuillastre, S.; Pauton, M.; Gao, L.; Desmarchelier, A.; Riives, A. J.; Prim, D.; Tondelier, D.; Geffroy, B.; Muller, G.; Clavier, G.; Pieters, G. J. Am. Chem. Soc. 2016, 138, 3990–3993. doi:10.1021/jacs.6b00850 |
| 21. | Hirai, H.; Nakajima, K.; Nakatsuka, S.; Shiren, K.; Ni, J.; Nomura, S.; Ikuta, T.; Hatakeyama, T. Angew. Chem., Int. Ed. 2015, 54, 13581–13585. doi:10.1002/anie.201506335 |
| 22. | Sasabe, H.; Hayasaka, Y.; Komatsu, R.; Nakao, K.; Kido, J. Chem. – Eur. J. 2017, 23, 114–119. doi:10.1002/chem.201604303 |
| 23. | Yao, L.; Zhang, S.; Wang, R.; Li, W.; Shen, F.; Yang, B.; Ma, Y. Angew. Chem., Int. Ed. 2014, 53, 2119–2123. doi:10.1002/anie.201308486 |
| 24. | Suzuki, K.; Kubo, S.; Shizu, K.; Fukushima, T.; Wakamiya, A.; Murata, Y.; Adachi, C.; Kaji, H. Angew. Chem., Int. Ed. 2015, 54, 15231–15235. doi:10.1002/anie.201508270 |
| 47. | Both the geometry optimization and energy calculation were performed by employing density functional theory (DFT), at the level of B3LYP/6-31G(d,p) on the Gaussian09 program package, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009. |
| 5. | Guliyev, R.; Coskun, A.; Akkaya, E. U. J. Am. Chem. Soc. 2009, 131, 9007–9013. doi:10.1021/ja902584a |
| 6. | Woodford, C. R.; Frady, E. P.; Smith, R. S.; Morey, B.; Canzi, G.; Palida, S. F.; Araneda, R. C.; Kristan, W. B., Jr.; Kubiak, C. P.; Miller, E. W.; Tsien, R. Y. J. Am. Chem. Soc. 2015, 137, 1817–1824. doi:10.1021/ja510602z |
| 7. | Escudero, D. Acc. Chem. Res. 2016, 49, 1816–1824. doi:10.1021/acs.accounts.6b00299 |
| 8. | Saha, M. L.; Yan, X.; Stang, P. J. Acc. Chem. Res. 2016, 49, 2527–2539. doi:10.1021/acs.accounts.6b00416 |
| 9. | Mahendran, V.; Pasumpon, K.; Thimmarayaperumal, S.; Thilagar, P.; Shanmugam, S. J. Org. Chem. 2016, 81, 3597–3602. doi:10.1021/acs.joc.6b00267 |
| 10. | Sandeep, A.; Praveen, V. K.; Kartha, K. K.; Karunakaran, V.; Ajayaghosh, A. Chem. Sci. 2016, 7, 4460–4467. doi:10.1039/c6sc00629a |
| 11. | Black, H. T.; Pelse, I.; Wolfe, R. M. W.; Reynolds, J. R. Chem. Commun. 2016, 52, 12877–12880. doi:10.1039/c6cc06443d |
| 12. | Ji, L.; Griesbeck, S.; Marder, T. B. Chem. Sci. 2017, 8, 846–863. doi:10.1039/c6sc04245g |
| 13. | Xu, Y.; Yu, S.; Chen, Q.; Chen, X.; Li, Y.; Yu, X.; Pu, L. Chem. – Eur. J. 2016, 22, 12061–12067. doi:10.1002/chem.201601540 |
| 14. | Zhou, J.; Outlaw, V. K.; Townsend, C. A.; Bragg, A. E. Chem. – Eur. J. 2016, 22, 15212–15215. doi:10.1002/chem.201603284 |
| 4. | Enoki, T.; Ohshita, J.; Ooyama, Y. Bull. Chem. Soc. Jpn. 2018, 91, 1704–1709. doi:10.1246/bcsj.20180210 |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 35. | Sumalekshmy, S.; Gopidas, K. R. J. Phys. Chem. B 2004, 108, 3705–3712. doi:10.1021/jp022549l |
| 36. | Sumalekshmy, S.; Gopidas, K. R. New J. Chem. 2005, 29, 325–331. doi:10.1039/b409411e |
| 37. | Sumalekshmy, S.; Gopidas, K. R. Photochem. Photobiol. Sci. 2005, 4, 539–546. doi:10.1039/b503251b |
| 38. | Dias, F. B.; Pollock, S.; Hedley, G.; Pålsson, L.-O.; Monkman, A.; Perepichka, I. I.; Perepichka, I. F.; Tavasli, M.; Bryce, M. R. J. Phys. Chem. B 2006, 110, 19329–19339. doi:10.1021/jp0643653 |
| 39. | Zhao, G.-J.; Chen, R.-K.; Sun, M.-T.; Liu, J.-Y.; Li, G.-Y.; Gao, Y.-L.; Han, K.-L.; Yang, X.-C.; Sun, L. Chem. – Eur. J. 2008, 14, 6935–6947. doi:10.1002/chem.200701868 |
| 40. | Aronica, C.; Venancio-Marques, A.; Chauvin, J.; Robert, V.; Lemercier, G. Chem. – Eur. J. 2009, 15, 5047–5055. doi:10.1002/chem.200802325 |
| 41. | Butler, R. S.; Cohn, P.; Tenzel, P.; Abboud, K. A.; Castellano, R. K. J. Am. Chem. Soc. 2009, 131, 623–633. doi:10.1021/ja806348z |
| 42. | Ooyama, Y.; Ito, G.; Kushimoto, K.; Komaguchi, K.; Imae, I.; Harima, Y. Org. Biomol. Chem. 2010, 8, 2756–2770. doi:10.1039/c003526b |
| 43. | Enoki, T.; Matsuo, K.; Ohshita, J.; Ooyama, Y. Phys. Chem. Chem. Phys. 2017, 19, 3565–3574. doi:10.1039/c6cp08573c |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 4. | Enoki, T.; Ohshita, J.; Ooyama, Y. Bull. Chem. Soc. Jpn. 2018, 91, 1704–1709. doi:10.1246/bcsj.20180210 |
| 44. | Lippert, E. Z. Naturforsch., A: Astrophys., Phys. Phys. Chem. 1955, 10, 541–545. doi:10.1515/zna-1955-0707 |
| 45. | Mataga, N.; Kaifu, Y.; Koizumi, M. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. doi:10.1246/bcsj.29.465 |
| 46. | Makitra, R. G. Reichardt, C., Solvents and Solvent Effects in Organic Chemistry, Weinheim: Wiley-VCH, 2003, 630 p.; Russian Journal of General Chemistry, Vol. 75; Springer Science and Business Media LLC, 2005; pp 664 ff. doi:10.1007/s11176-005-0294-y |
| 2. | Ooyama, Y.; Uenaka, K.; Ohshita, J. Eur. J. Org. Chem. 2015, 3713–3720. doi:10.1002/ejoc.201500341 |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 3. | Ooyama, Y.; Uenaka, K.; Harima, Y.; Ohshita, J. RSC Adv. 2014, 4, 30225–30228. doi:10.1039/c4ra03999h |
| 4. | Enoki, T.; Ohshita, J.; Ooyama, Y. Bull. Chem. Soc. Jpn. 2018, 91, 1704–1709. doi:10.1246/bcsj.20180210 |
| 1. | Ooyama, Y.; Uenaka, K.; Ohshita, J. RSC Adv. 2015, 5, 21012–21018. doi:10.1039/c4ra16399k |
© 2019 Imato et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)