Abstract
Triangulenium dyes functionalized with one, two or three ethylthiol functionalities were synthesized and their optical properties were studied. The sulfur functionalities were introduced by aromatic nucleophilic substitution of methoxy groups in triarylmethylium cations with ethanethiol followed by partial or full ring closure of the ortho positions with nitrogen or oxygen bridges leading to sulfur-functionalized acridinium, xanthenium or triangulenium dyes. For all the dye classes the sulfur functionalities are found to lead to intensely absorbing dyes in the visible range (470 to 515 nm), quite similar to known analogous dye systems with dialkylamino donor groups in place of the ethylthiol substituents. For the triangulenium derivatives significant fluorescence was observed (Φf = 0.1 to Φf = 0.3).

Graphical Abstract
Introduction
The design, synthesis and studies of organic fluorescent dyes have witnessed a revival in recent years, in particular due to their applications in imaging and biomedical assays and analytical techniques [1-5]. The desire to detect minute amounts of dye, ideally single molecules [6,7], in complex biological environments with high levels of autofluorescence, constantly challenges chemists to develop new dyes with improved or special properties. In the design of simple dyes parameters such as molar absorption coefficients (ε), absorption/emission wavelengths [8,9], fluorescence quantum yields (Φfl) [10,11], and fluorescence lifetime (τfl) [12,13] are key photophysical properties to consider and optimize for any given application.
We have for quite some time been interested in the synthesis, properties and applications of dyes from the triangulenium family (Figure 1) [14,15]. The triangulenium dyes can be divided into two main categories: 1) triangulenium dyes with donor substituents at the corners of the triangulenium ring system (position 2, 6 and 10, Figure 1a) [16-18], and 2) triangulenium dyes without such groups (Figure 1b) [19-21]. Dyes in the first category have intense absorption (ε ≈ 50,000–130,000 M−1·cm−1), high fluorescence quantum yields (Φfl > 50%) and fluorescence lifetimes of 3–4 ns. All properties that agree well with their structural resemblance to rhodamines and fluoresceines, and triangulenium dyes such as A3-TOTA+ and H-TOTA+ (Figure 1a) can be viewed as extended symmetric versions of these prominent dyes [16,22]. The second class of triangulenium dyes, without appended donor groups, are characterized by much less intense transitions (ε ≈ 5,000–20,000 M−1·cm−1), which for some derivatives leads to unusually long fluorescence lifetimes (τfl ≈ 20 ns) [23,24]. This long fluorescence lifetime has been a key point of interest since it enables time-gated detection for suppression of autofluorescence [25,26] and provides attractive advantages in fluorescence polarization assays [13,27,28].
![[1860-5397-15-210-1]](/bjoc/content/figures/1860-5397-15-210-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of some representative triangulenium dyes. a) Rhodamine/fluorescine-like derivatives with donor groups in para-positions (2, 6, and 10) to the formal cation center (12c). b) Derivatives without donor groups.
Figure 1: Structures of some representative triangulenium dyes. a) Rhodamine/fluorescine-like derivatives wit...
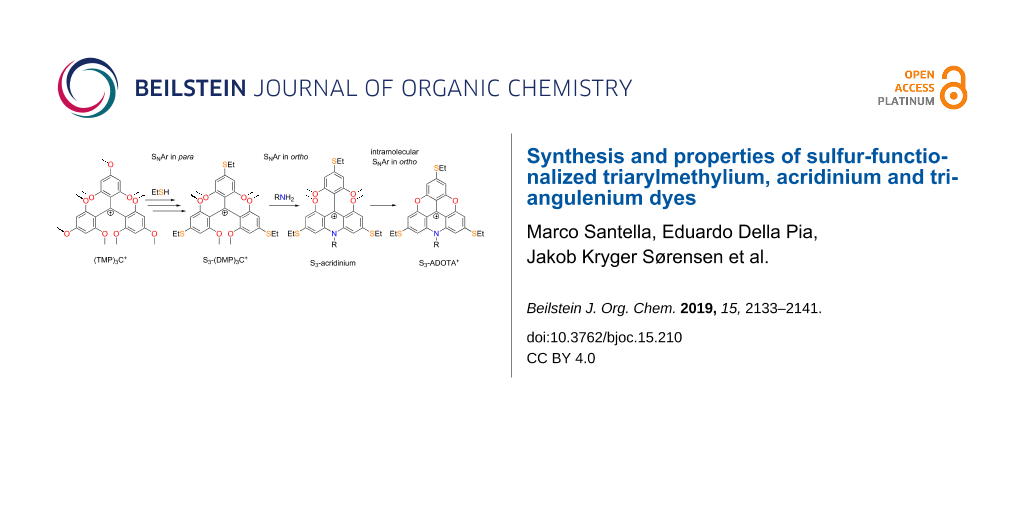
A common characteristic feature of triangulenium dye synthesis is the use of methoxy-substituted triarylmethylium salts as simple precursors allowing both introduction of dialkylamino donor groups and formation of the heterocyclic triangulenium ring systems. These characteristic types of aromatic nucleophilic substitution (SNAr) reactions are exemplified by the synthesis of A3-ADOTA+ (Figure 2) [17]. Starting from the readily available tris(2,4,6-trimethoxyphenyl)methylium salt (TMP)3C+ [18,29], stepwise replacement of the para-methoxy groups by dialkylamines provides access to a wide variety of symmetric and asymmetric triarylmethylium dyes [18,30,31]. Replacement of two o-methoxy groups by one primary amine gives acridinium-type ring systems (Figure 2, step 2) and is a key reaction for the formation of the unsubstituted triangulenium dyes shown in Figure 1b [19,20]. Finally, formation of oxygen bridges in the triangulenium system (Figure 2, step 3) involves demethylation conditions and intramolecular SNAr replacement of ortho-methoxy groups [18,32].
![[1860-5397-15-210-2]](/bjoc/content/figures/1860-5397-15-210-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Examples of various types of SNAr reactions typical in triangulenium synthesis, exemplified with the synthesis of A3-ADOTA+: step 1, replacement of p-methoxy groups with dialkylamines. Step 2, replacement of o-methoxy groups with a primary amine followed by intramolecular SNAr reaction. Step 3, intramolecular SNAr replacement of methoxy groups by hydroxy groups formed under ether-cleavage conditions.
Figure 2: Examples of various types of SNAr reactions typical in triangulenium synthesis, exemplified with th...
The SNAr approach to the synthesis of triangulenium dyes [14,18,19] has been extremely successful and expanded the family greatly from the single derivative (TOTA+, Figure 1b) first synthesized by Martin and Smith in 1964 [32], and also includes the family of helicenium dyes [33-35]. However, the introduction of groups other than nitrogen and oxygen has so far not been performed by the SNAr approach. Thus in the preparation of the sulfur-bridged triangulenium ions DOTTA+ and AOTTA+ (Figure 1b) Lacour and co-workers reported unsuccessful attempts of SNAr reactions with sulfur nucleophiles in ortho-position of (TMP)3C+ and had to assemble the thioxanthenium part of the triangulenium ring system independently by other means [36]. Similarly, we had to use a stepwise buildup of the triangulenium systems to introduce saturated [37] and unsaturated [38] carbon bridges.
Here we report for the first time the introduction of sulfur functionalities into triangulenium dyes by SNAr reaction with ethylthiol nucleophiles in the para-positions accessing several new families of xanthenium, acridinium and triangulenium dyes with thioether donor groups.
Results and Discussion
Firstly, a series of triarylmethylium salts with variable number of para-methoxy substituents was synthesized. The easily achievable cations (TMP)3C+, (DMP)(TMP)2C+ and (DMP)2(TMP)C+ (Scheme 1) were prepared by their respective literature procedures [18,31]. To investigate the reactivity of these carbenium systems in SNAr reactions with sulfur-based nucleophiles, simple alkylthiols were chosen, with the ethyl and tert-butyl thiols being the primary choice. SNAr reactions with the two thiols were tested under identical reaction conditions (Scheme 1).
![[1860-5397-15-210-i1]](/bjoc/content/inline/1860-5397-15-210-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of three novel SX-(DMP)3C+ PF6− ethylsulfanyl-substituted triarylmethylium salts.
Scheme 1: Synthesis of three novel SX-(DMP)3C+ PF6− ethylsulfanyl-substituted triarylmethylium salts.
These conditions consisted of heating the reaction components in refluxing acetonitrile in the presence of collidine as base. For all three carbenium salts examinations showed that only ethanethiol lead to substitution. The progress of the reaction was conveniently followed by MALDI–TOF mass spectrometry. In case of the reactions with tert-butylthiolate, neither detection of the target molecule nor any of the intermediates were observed. This lack of reactivity is likely explained by the tert-butylthiolate nucleophile being too bulky to undergo reaction. In the successful reactions, which had occurred with ethanethiol, a high selectivity was observed for para-substitutions, giving Sx-(DMP)3C+ 1, 2, and 3 in reasonable yields of 20–50% after column chromatography purification. It is important to note that the gradual introduction of thioethers into the carbenium systems did not significantly influence the overall reactivity of the system towards subsequent nucleophilic aromatic substitution. When the reaction was followed by MALDI–TOFMS spectrometry it was thus possible to observe simultaneously the presence of the target compound and all of the intermediates involved in the reaction. This behavior is contrary to the reaction pattern observed when using dialkylamines as nucleophiles, where the strong electron-donating effect of the introduced amines stabilize the carbenium ion products and thus significantly reduces the reactivity of the remaining methoxy groups for further substitutions [18,39]. This observation is in agreement with the much stronger cation stabilization of the dialkylamino group compared to the methoxy group. The ability of the alkylthio group to stabilize carbenium ions, given by the Hammet σp+ value [40], on the other hand is quite similar to the methoxy group or even a little lower [41], and does thus not slow down the multistep SNAr reactions.
The new ortho-methoxytriarylcarbenium ions with one, two and three para-SEt groups (1–3) are potential precursors for a wide variety of new triangulenium, xanthenium, and acridinium dyes. To elucidate some of these possibilities we first investigated transformations of the symmetric derivative 1. Treatment with primary alkylamines, n-propylamine and n-octylamine, yielded exclusively the acridinium products 4a,b (Scheme 2). This ortho SNAr transformation is similar to what is reported for the (DMP)3C+ system [19,20,42] lacking para-substituents and for the para-amino-substituted analogue [17] (step 2 in Figure 1). It was found that the reactivity in SNAr reactions of 1 with primary amines was high and the acridinium compounds 4a,b were obtained in few minutes after the addition of 2 equiv of the corresponding primary amine at room temperature. Further ring closure to two oxygen bridges in acridinium compounds 4a and 4b to the corresponding triangulenium dyes S3-ADOTA+ (5a,b) was achieved by heating in molten pyridine hydrochloride (Scheme 2).
![[1860-5397-15-210-i2]](/bjoc/content/inline/1860-5397-15-210-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic route for the synthesis of S3-ADOTA+.
Scheme 2: Synthetic route for the synthesis of S3-ADOTA+.
It is noteworthy that the ethylthio ether linkages remained intact upon treatment with molten PyrHCl, which was found to result in complicated mixtures of dealkylated byproducts when these conditions were applied on dialkylamino-substituted carbenium systems [18].
The direct ring closure of 1 in PyrHCl yielded in a similar manner the sulfur-functionalized trioxatriangulenium system S3-TOTA+ (6) as shown in Scheme 3. Mono ring closure was achieved under milder ether cleaving conditions with aqueous HBr in acetic acid, leading to the ethylthio-substituted xanthenium system 7 in 43% yield (Scheme 3).
![[1860-5397-15-210-i3]](/bjoc/content/inline/1860-5397-15-210-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of S3-TOTA+ PF6− (6) and the mono ring closed xanthenium 7.
Scheme 3: Synthesis of S3-TOTA+ PF6− (6) and the mono ring closed xanthenium 7.
By applying similar molten pyridine hydrochloride conditions to the mono- and disubstituted thioether carbenium salts (2 and 3), it was possible to isolate the derivatives S2-TOTA+ (8) and S1-TOTA+ (9), respectively as their hexafluorophosphate salts (Scheme 4). The two Sx-TOTA+ derivatives were obtained with good yield after purification by column chromatography and subsequent recrystallization.
![[1860-5397-15-210-i4]](/bjoc/content/inline/1860-5397-15-210-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of S2-TOTA+ PF6− (8) and S1-TOTA+ PF6− (9).
Scheme 4: Synthesis of S2-TOTA+ PF6− (8) and S1-TOTA+ PF6− (9).
To conclude, the successful introduction of -SEt groups by the SNAr approach, and subsequent nitrogen and oxygen ring-closure reactions provides access to several new families of carbenium dyes, all with the unusual -SR donor group: thus 1–3 represent new triarylmethylium dyes, 4a and 4b sulfur analogues of aminoacridinium dyes (acridine orange-like structures), 7 a fluorescein-like xanthenium dye, 5a and 5b are sulfur-substituted ADOTA+ dyes, and finally the three sulfur-substituted TOTA+ dyes 6, 8 and 9.
Now the relevant questions are: how do the -SR donor groups influence transition energies and intensities? And how do they affect fluorescence quantum yields in these new dye systems? To the extent possible we will compare the new sulfur-functionalized dyes to known analogues with -OR or -NR2 donor groups in the same positions.
The sulfur-substituted triarylmethylium dyes 1, 2 and 3 display broad absorption bands in the 500–700 nm region (Figure 3), that in shape and relative transition energy are quite similar to the analogues with similar numbers of para-methoxy or diethylamino groups [31], as shown by comparison of maximum absorption wavelength (λmax,abs) and molar absorptivity (ε) in Table 1. It is noticed that the -SEt donor group in these ortho-hexamethoxytriarylmethylium dyes provides transition energies and intensities very similar to those of commonly used dialkylamino-donor groups, but significantly red-shifted relative to the methoxy-substituted analogues.
![[1860-5397-15-210-3]](/bjoc/content/figures/1860-5397-15-210-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra in MeCN: S3-(DMP)3C+ (1, red), S2-(DMP)3C+ (2, green), and S1-(DMP)3C+ (3, blue).
Figure 3: UV–vis spectra in MeCN: S3-(DMP)3C+ (1, red), S2-(DMP)3C+ (2, green), and S1-(DMP)3C+ (3, blue).
Table 1: Summary of absorption data of triarylmethylium ions in MeCN.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-210-i5.png?max-width=637&scale=1.0)
|
|||
| λmax.abs. (ε, M−1·cm−1) | |||
|
donor groups
R1, R2, R3 |
-SEt | -OMea | -NEt2a |
|
one donor
R2 = R3 = H |
576 nm
(24600) |
491 nm
(14100) |
457 nm
(16900) |
|
two donors
R3 = H |
639 nm
(34600) |
580 nm
(18400) |
637 nm
(40400) |
| three donors |
642 nm
(49800) |
577 nm
(23400) |
634 nm
(49400) |
aData from [43,44].
Absorption spectra of the partially ring-closed acridinium and xanthenium compounds, with three para-SEt groups, 4a and 7, respectively, are shown in Figure 4. For these compounds the spectra are dominated by strong transitions assigned to the 3,6-diethylthio-acridinium and -xanthenium ring systems peaking at 457 nm (ε = 47000 M−1·cm−1) and at 520 nm (ε = 60000 M−1·cm−1), respectively (see Supporting Information File 1, Table S1 for additional data in more solvents). The energy and intensity of these transitions are quite similar to those found in dialkylamino analogues, that are 3,6-diaminoacridines and rhodamines [43,44]. The weak tails on the red side of these bands are tentatively assigned to internal charge-transfer transitions from the perpendicularly [19,42] arranged ethylthio(dimethoxy)phenyl group to the xanthenium/acridinium systems polarized along the y-axes (Figure 4, inset). This bichromophoric behavior has been studied in detail for the dialkylamino-substituted xanthenium/rhodamine system [45,46], and is also the likely reason for these compounds being non-fluorescent.
![[1860-5397-15-210-4]](/bjoc/content/figures/1860-5397-15-210-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis spectra in MeCN: S3-acridinium (4a, black) and S3-xanthenium (7, red). Inset: The 3D structure of 4a with indication of the principle axes of the electronic transitions.
Figure 4: UV–vis spectra in MeCN: S3-acridinium (4a, black) and S3-xanthenium (7, red). Inset: The 3D structu...
The three sulfur-substituted trioxatriangulenium dyes 6, 8, and 9 all display a first absorption band around 480 nm (Figure 5), with increasing intensity as the number of -SEt groups on the TOTA+ core increases. This behavior resembles the trend observed for the analogue series of amino-substituted TOTA’s (Table 2) [18,31]. In the two low-symmetry derivatives 8 and 9 transitions to the S2 excited states are observed at around 400 nm, while the D3h symmetric S3-TOTA+ shows only one, though broad, absorption band corresponding to merging of the S1 and S2 transitions into one, arising from the degenerated HOMO in the symmetric dye. The influence of solvent and counter ions on such degenerate states have been studied in detail for the A3-TOTA system [22,47] and related triarylmethylium dyes such as crystal violet [48,49].
![[1860-5397-15-210-5]](/bjoc/content/figures/1860-5397-15-210-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: UV–vis spectra in CH2Cl2: S1-TOTA+ (9, blue line), S2-TOTA+ (8, red line), and S3-TOTA+ (6, black line).
Figure 5: UV–vis spectra in CH2Cl2: S1-TOTA+ (9, blue line), S2-TOTA+ (8, red line), and S3-TOTA+ (6, black l...
Table 2: Summary of absorption data of substituted TOTA dyes in CH2Cl2.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-210-i6.png?max-width=637&scale=1.0)
|
||
| λmax,abs (ε, M−1·cm−1) | ||
|
Donor groups
R1, R2, R3 |
-SEt | -NEt2 |
|
one donor
R2 = R3 = H |
S1-TOTA+ (9)
483 nm (35000) |
A1-TOTA+a
507 nm (41700) |
|
two donors
R3 = H |
S2-TOTA+ (8)
487 nm (65200) |
A2-TOTA+a
513 nm (59700) |
| three donors |
S3-TOTA+ (6)
478 nm (91900) |
A3-TOTA+b
471 nm (132900) |
When three -SEt groups are placed on the asymmetric azadioxatriangulenium core, as in S3-ADOTA+ (5a), the presence of two electronic transitions becomes very clear, with two well-resolved peaks in the absorption spectrum (Figure 6). The transition at 442 nm is assigned to the S0 → S2 transition and nearly coincides with the main transition observed in the S3-acridinium (4a) precursor before ring closure (Figure 4), indicating that this, the most intense transition belongs to the same chromophore, now part of the triangulenium ring system. The S0 → S1 transition in 5a is found at 507 nm, where the open form only had a very weak shoulder in its absorption spectrum (Figure 4). The ring closure of 4a into the fully planar triangulenium system 5a leads to a significant increase in the orbital overlap and thus also in the intensity of the S0 → S1 transition. This assignment is supported by calculations of the orbitals involved in the first two electronic transitions (Figure 6), which confirm their localization in different parts of the ADOTA+ system. The much more allowed S0 → S1 transition is also in agreement with the observation that 5a (and 5b) display intense fluorescence (Figure 6).
![[1860-5397-15-210-6]](/bjoc/content/figures/1860-5397-15-210-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: UV–vis absorption and fluorescence spectra (λex = 485 nm) of 5a in CH2Cl2 solution. Calculated molecular orbital contour plots (semi-empirical method AM1).
Figure 6: UV–vis absorption and fluorescence spectra (λex = 485 nm) of 5a in CH2Cl2 solution. Calculated mole...
Table 3 summarizes the spectral and photophysical properties on the triangulenium dyes showing any applicable fluorescence. Beside S3-ADOTA+ (5a) that are the double and triple -SEt-substituted TOTAs 6 and 8, for which the fluorescence spectra are shown in Figure 7, with fluorescence quantum yields of 16% and 12%, respectively. From the measured fluorescence lifetimes and quantum yields (Table 3) it is possible to calculate the radiative lifetimes (τ0), which are found to be in qualitative agreement with the molar absorption coefficients (ε) for the corresponding transitions, as expected from the Strickler–Berg relation [50].
Table 3: Summary of optical properties of the fluorescent derivatives.
| Compound | Solvent | λmax,abs (nm) | ε (M−1·cm−1) | λmax,fl (nm) | Φfa | τ (ns) | τ0b (ns) |
| 5a (S3-ADOTA) | CH2Cl2 |
442
507 |
76700
35400 |
532 | 0.28 | 3.9 | 13.9 |
| 6 (S3-TOTA) | CH2Cl2 | 478 | 91850 | 505 | 0.16 | 0.7 | 4.4 |
| 8 (S2-TOTA) | CH2Cl2 | 487 | 65200 | 509 | 0.12 | 0.7 | 5.8 |
aMeasured relative to fluorescein in 0.1 M aqueous NaOH (Φ = 0.96); bradiative lifetime τ0 = Φf/τ.
![[1860-5397-15-210-7]](/bjoc/content/figures/1860-5397-15-210-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Normalized absorption and fluorescence spectra of 6 (S3-TOTA+), λex = 460 nm, and 8 (S2-TOTA+), λex = 470 nm, in CH2Cl2 solution.
Figure 7: Normalized absorption and fluorescence spectra of 6 (S3-TOTA+), λex = 460 nm, and 8 (S2-TOTA+), λex...
While the spectral properties of the new -SEt-substituted dyes are surprisingly similar to the -NEt2-substituted analogues across the various dye families they are obviously less bright fluorophores. Thus, the dialkylamino-substituted analogue of 5a (A3-ADOTA+) has a reported quantum yield as high as 64% in acetonitrile [17], on par with A3-TOTA+ and A2-TOTA+ which display quantum yields from 50–100% depending on the solvent [31]. A similar reduction in fluorescence efficiency was observed by Kotaskova et al. for fluorescein derivatives with one alkylthio group in the 3 position replacing an -OH/-O− group [51]. The origin of reduced fluorescence quantum yields in dyes with alkylthio donor groups in their chromophores is not clear at this point. It may result from enhanced internal conversion or intersystem crossing to the triplet state. Further photophysical work will have to settle this issue and thereby suggest structural improvements and/or the best applications of these dyes.
Conclusion
The effective introduction of alkylthiol groups into the para-positions of triarylmethylium ions via SNAr reactions was demonstrated. These new thioether-substituted triarylmethylium ions provide access to a broad range of new heterocyclic carbenium dyes of the xanthenium, acridinium and triangulenium type via further SNAr reactions with primary amines and ring-closure reactions. The introduction of thioether donor groups in these dye classes is unprecedented, but is found to yield spectral properties quite similar to analogous dyes with dialkylamino groups. The synthesized thioether-substituted triangulenium derivatives are fluorescent, though with lower quantum yields (Φf = 0.1 to Φf = 0.3) than the corresponding dialkylamino-substituted analogues.
Supporting Information
| Supporting Information File 1: Experimental details, full synthetic procedures, spectroscopic characterization and NMR spectra of new compounds, as well as additional UV–vis and fluorescence spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Lavis, L. D.; Raines, R. T. ACS Chem. Biol. 2008, 3, 142–155. doi:10.1021/cb700248m
Return to citation in text: [1] -
Grimm, J. B.; Muthusamy, A. K.; Liang, Y.; Brown, T. A.; Lemon, W. C.; Patel, R.; Lu, R.; Macklin, J. J.; Keller, P. J.; Ji, N.; Lavis, L. D. Nat. Methods 2017, 14, 987–994. doi:10.1038/nmeth.4403
Return to citation in text: [1] -
Butkevich, A. N.; Lukinavičius, G.; D’Este, E.; Hell, S. W. J. Am. Chem. Soc. 2017, 139, 12378–12381. doi:10.1021/jacs.7b06412
Return to citation in text: [1] -
Kolmakov, K.; Hebisch, E.; Wolfram, T.; Nordwig, L. A.; Wurm, C. A.; Ta, H.; Westphal, V.; Belov, V. N.; Hell, S. W. Chem. – Eur. J. 2015, 21, 13344–13356. doi:10.1002/chem.201501394
Return to citation in text: [1] -
Haugland, R. P. The Molecular Probes Handbook, 11th ed.; Thermofischer Scientific, 2010.
Return to citation in text: [1] -
Moerner, W. E.; Orrit, M. Science 1999, 283, 1670–1676. doi:10.1126/science.283.5408.1670
Return to citation in text: [1] -
Grimm, J. B.; English, B. P.; Chen, J.; Slaughter, J. P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J. J.; Normanno, D.; Singer, R. H.; Lionnet, T.; Lavis, L. D. Nat. Methods 2015, 12, 244–250. doi:10.1038/nmeth.3256
Return to citation in text: [1] -
Lei, Z.; Li, X.; Luo, X.; He, H.; Zheng, J.; Qian, X.; Yang, Y. Angew. Chem., Int. Ed. 2017, 56, 2979–2983. doi:10.1002/anie.201612301
Return to citation in text: [1] -
Staudinger, C.; Borisov, S. M. Methods Appl. Fluoresc. 2015, 3, 042005. doi:10.1088/2050-6120/3/4/042005
Return to citation in text: [1] -
Panchuk-Voloshina, N.; Haugland, R. P.; Bishop-Stewart, J.; Bhalgat, M. K.; Millard, P. J.; Mao, F.; Leung, W.-Y.; Haugland, R. P. J. Histochem. Cytochem. 1999, 47, 1179–1188. doi:10.1177/002215549904700910
Return to citation in text: [1] -
Song, X.; Johnson, A.; Foley, J. J. Am. Chem. Soc. 2008, 130, 17652–17653. doi:10.1021/ja8075617
Return to citation in text: [1] -
Berezin, M. Y.; Achilefu, S. Chem. Rev. 2010, 110, 2641–2684. doi:10.1021/cr900343z
Return to citation in text: [1] -
Meyer-Almes, F.-J. Methods Appl. Fluoresc. 2017, 5, 042002. doi:10.1088/2050-6120/aa7c7a
Return to citation in text: [1] [2] -
Bosson, J.; Gouin, J.; Lacour, J. Chem. Soc. Rev. 2014, 43, 2824–2840. doi:10.1039/c3cs60461f
Return to citation in text: [1] [2] -
Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076
Return to citation in text: [1] -
Westerlund, F.; Hildebrandt, C. B.; Sørensen, T. J.; Laursen, B. W. Chem. – Eur. J. 2010, 16, 2992–2996. doi:10.1002/chem.200902965
Return to citation in text: [1] [2] -
Laursen, B. W.; Sørensen, T. J. J. Org. Chem. 2009, 74, 3183–3185. doi:10.1021/jo9002486
Return to citation in text: [1] [2] [3] [4] -
Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f
Return to citation in text: [1] [2] [3] [4] [5] -
Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s
Return to citation in text: [1] [2] [3] -
Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732
Return to citation in text: [1] -
Laursen, B. W.; Reynisson, J.; Mikkelsen, K. V.; Bechgaard, K.; Harrit, N. Photochem. Photobiol. Sci. 2005, 4, 568–576. doi:10.1039/b501584g
Return to citation in text: [1] [2] -
Bogh, S. A.; Simmermacher, M.; Westberg, M.; Bregnhøj, M.; Rosenberg, M.; De Vico, L.; Veiga, M.; Laursen, B. W.; Ogilby, P. R.; Sauer, S. P. A.; Sørensen, T. J. ACS Omega 2017, 2, 193–203. doi:10.1021/acsomega.6b00211
Return to citation in text: [1] -
Bogh, S. A.; Bora, I.; Rosenberg, M.; Thyrhaug, E.; Laursen, B. W.; Sørensen, T. J. Methods Appl. Fluoresc. 2015, 3, 045001. doi:10.1088/2050-6120/3/4/045001
Return to citation in text: [1] -
Rich, R. M.; Stankowska, D. L.; Maliwal, B. P.; Sørensen, T. J.; Laursen, B. W.; Krishnamoorthy, R. R.; Gryczynski, Z.; Borejdo, J.; Gryczynski, I.; Fudala, R. Anal. Bioanal. Chem. 2013, 405, 2065–2075. doi:10.1007/s00216-012-6623-1
Return to citation in text: [1] -
Rich, R. M.; Mummert, M.; Gryczynski, Z.; Borejdo, J.; Sørensen, T. J.; Laursen, B. W.; Foldes-Papp, Z.; Gryczynski, I.; Fudala, R. Anal. Bioanal. Chem. 2013, 405, 4887–4894. doi:10.1007/s00216-013-6879-0
Return to citation in text: [1] -
Hall, M. D.; Yasgar, A.; Peryea, T.; Braisted, J. C.; Jadhav, A.; Simeonov, A.; Coussens, N. P. Methods Appl. Fluoresc. 2016, 4, 022001. doi:10.1088/2050-6120/4/2/022001
Return to citation in text: [1] -
Sørensen, T. J.; Thyrhaug, E.; Szabelski, M.; Luchowski, R.; Gryczynski, I.; Gryczynski, Z.; Laursen, B. W. Methods Appl. Fluoresc. 2013, 1, 025001. doi:10.1088/2050-6120/1/2/025001
Return to citation in text: [1] -
Wada, M.; Konishi, H.; Kirishima, K.; Takeuchi, H.; Natsume, S.; Erabi, T. Bull. Chem. Soc. Jpn. 1997, 70, 2737–2741. doi:10.1246/bcsj.70.2737
Return to citation in text: [1] -
Laursen, B. W.; Nørgaard, K.; Reitzel, N.; Simonsen, J. B.; Nielsen, C. B.; Als-Nielsen, J.; Bjørnholm, T.; Sølling, T. I.; Nielsen, M. M.; Bunk, O.; Kjaer, K.; Tchebotareva, N.; Watson, M. D.; Müllen, K.; Piris, J. Langmuir 2004, 20, 4139–4146. doi:10.1021/la049944i
Return to citation in text: [1] -
Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Martin, J. C.; Smith, R. G. J. Am. Chem. Soc. 1964, 86, 2252–2256. doi:10.1021/ja01065a030
Return to citation in text: [1] [2] -
Laleu, B.; Mobian, P.; Herse, C.; Laursen, B. W.; Hopfgartner, G.; Bernardinelli, G.; Lacour, J. Angew. Chem., Int. Ed. 2005, 44, 1879–1883. doi:10.1002/anie.200462321
Return to citation in text: [1] -
Herse, C.; Bas, D.; Krebs, F. C.; Bürgi, T.; Weber, J.; Wesolowski, T.; Laursen, B. W.; Lacour, J. Angew. Chem., Int. Ed. 2003, 42, 3162–3166. doi:10.1002/anie.200351443
Return to citation in text: [1] -
Bosson, J.; Labrador, G. M.; Pascal, S.; Miannay, F.-A.; Yushchenko, O.; Li, H.; Bouffier, L.; Sojic, N.; Tovar, R. C.; Muller, G.; Jacquemin, D.; Laurent, A. D.; Le Guennic, B.; Vauthey, E.; Lacour, J. Chem. – Eur. J. 2016, 22, 18394–18403. doi:10.1002/chem.201603591
Return to citation in text: [1] -
Nicolas, C.; Bernardinelli, G.; Lacour, J. J. Phys. Org. Chem. 2010, 23, 1049–1056. doi:10.1002/poc.1753
Return to citation in text: [1] -
Rosenberg, M.; Rostgaard, K. R.; Liao, Z.; Madsen, A. Ø.; Martinez, K. L.; Vosch, T.; Laursen, B. W. Chem. Sci. 2018, 9, 3122–3130. doi:10.1039/c8sc00089a
Return to citation in text: [1] -
Rosenberg, M.; Santella, M.; Bogh, S. A.; Muñoz, A. V.; Andersen, H. O. B.; Hammerich, O.; Bora, I.; Lincke, K.; Laursen, B. W. J. Org. Chem. 2019, 84, 2556–2567. doi:10.1021/acs.joc.8b02978
Return to citation in text: [1] -
Simonsen, J. B.; Kjær, K.; Howes, P.; Nørgaard, K.; Bjørnholm, T.; Harrit, N.; Laursen, B. W. Langmuir 2009, 25, 3584–3592. doi:10.1021/la803733s
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Brown, H. C.; Okamoto, Y. J. Am. Chem. Soc. 1958, 80, 4979–4987. doi:10.1021/ja01551a055
Return to citation in text: [1] -
Laleu, B.; Herse, C.; Laursen, B. W.; Bernardinelli, G.; Lacour, J. J. Org. Chem. 2003, 68, 6304–6308. doi:10.1021/jo0345998
Return to citation in text: [1] [2] -
Drexhage, K. H. Structure and Properties of Laser Dyes. In Dye Lasers; Schäfer, F. P., Ed.; Topics in Applied Physics, Vol. 1; Springer: Berlin, Heidelberg, Germany, 1973; pp 144–193. doi:10.1007/978-3-662-11579-4_4
Return to citation in text: [1] [2] -
Griffiths, J. Colour and Constitution of Organic Molecules; Academic Press: London, 1976.
Return to citation in text: [1] [2] -
Sørensen, T. J.; Kilså, K.; Laursen, B. W. Chem. – Eur. J. 2015, 21, 8521–8529. doi:10.1002/chem.201500355
Return to citation in text: [1] -
Sørensen, T. J.; Shi, D.; Laursen, B. W. Chem. – Eur. J. 2016, 22, 7046–7049. doi:10.1002/chem.201600496
Return to citation in text: [1] -
Westerlund, F.; Elm, J.; Lykkebo, J.; Carlsson, N.; Thyrhaug, E.; Åkerman, B.; Sørensen, T. J.; Mikkelsen, K. V.; Laursen, B. W. Photochem. Photobiol. Sci. 2011, 10, 1963–1973. doi:10.1039/c1pp05253e
Return to citation in text: [1] -
Lueck, H. B.; McHale, J. L.; Edwards, W. D. J. Am. Chem. Soc. 1992, 114, 2342–2348. doi:10.1021/ja00033a007
Return to citation in text: [1] -
Lewis, L. M.; Indig, G. L. Dyes Pigm. 2000, 46, 145–154. doi:10.1016/s0143-7208(00)00049-8
Return to citation in text: [1] -
Strickler, S. J.; Berg, R. A. J. Chem. Phys. 1962, 37, 814–822. doi:10.1063/1.1733166
Return to citation in text: [1] -
Kotaskova, M.; Osman Oglou, O.; Helm, M. Org. Biomol. Chem. 2014, 12, 3816–3820. doi:10.1039/c4ob00533c
Return to citation in text: [1]
| 19. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 20. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 42. | Laleu, B.; Herse, C.; Laursen, B. W.; Bernardinelli, G.; Lacour, J. J. Org. Chem. 2003, 68, 6304–6308. doi:10.1021/jo0345998 |
| 17. | Laursen, B. W.; Sørensen, T. J. J. Org. Chem. 2009, 74, 3183–3185. doi:10.1021/jo9002486 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 1. | Lavis, L. D.; Raines, R. T. ACS Chem. Biol. 2008, 3, 142–155. doi:10.1021/cb700248m |
| 2. | Grimm, J. B.; Muthusamy, A. K.; Liang, Y.; Brown, T. A.; Lemon, W. C.; Patel, R.; Lu, R.; Macklin, J. J.; Keller, P. J.; Ji, N.; Lavis, L. D. Nat. Methods 2017, 14, 987–994. doi:10.1038/nmeth.4403 |
| 3. | Butkevich, A. N.; Lukinavičius, G.; D’Este, E.; Hell, S. W. J. Am. Chem. Soc. 2017, 139, 12378–12381. doi:10.1021/jacs.7b06412 |
| 4. | Kolmakov, K.; Hebisch, E.; Wolfram, T.; Nordwig, L. A.; Wurm, C. A.; Ta, H.; Westphal, V.; Belov, V. N.; Hell, S. W. Chem. – Eur. J. 2015, 21, 13344–13356. doi:10.1002/chem.201501394 |
| 5. | Haugland, R. P. The Molecular Probes Handbook, 11th ed.; Thermofischer Scientific, 2010. |
| 12. | Berezin, M. Y.; Achilefu, S. Chem. Rev. 2010, 110, 2641–2684. doi:10.1021/cr900343z |
| 13. | Meyer-Almes, F.-J. Methods Appl. Fluoresc. 2017, 5, 042002. doi:10.1088/2050-6120/aa7c7a |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 30. | Laursen, B. W.; Nørgaard, K.; Reitzel, N.; Simonsen, J. B.; Nielsen, C. B.; Als-Nielsen, J.; Bjørnholm, T.; Sølling, T. I.; Nielsen, M. M.; Bunk, O.; Kjaer, K.; Tchebotareva, N.; Watson, M. D.; Müllen, K.; Piris, J. Langmuir 2004, 20, 4139–4146. doi:10.1021/la049944i |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
| 22. | Laursen, B. W.; Reynisson, J.; Mikkelsen, K. V.; Bechgaard, K.; Harrit, N. Photochem. Photobiol. Sci. 2005, 4, 568–576. doi:10.1039/b501584g |
| 47. | Westerlund, F.; Elm, J.; Lykkebo, J.; Carlsson, N.; Thyrhaug, E.; Åkerman, B.; Sørensen, T. J.; Mikkelsen, K. V.; Laursen, B. W. Photochem. Photobiol. Sci. 2011, 10, 1963–1973. doi:10.1039/c1pp05253e |
| 10. | Panchuk-Voloshina, N.; Haugland, R. P.; Bishop-Stewart, J.; Bhalgat, M. K.; Millard, P. J.; Mao, F.; Leung, W.-Y.; Haugland, R. P. J. Histochem. Cytochem. 1999, 47, 1179–1188. doi:10.1177/002215549904700910 |
| 11. | Song, X.; Johnson, A.; Foley, J. J. Am. Chem. Soc. 2008, 130, 17652–17653. doi:10.1021/ja8075617 |
| 19. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 20. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 48. | Lueck, H. B.; McHale, J. L.; Edwards, W. D. J. Am. Chem. Soc. 1992, 114, 2342–2348. doi:10.1021/ja00033a007 |
| 49. | Lewis, L. M.; Indig, G. L. Dyes Pigm. 2000, 46, 145–154. doi:10.1016/s0143-7208(00)00049-8 |
| 8. | Lei, Z.; Li, X.; Luo, X.; He, H.; Zheng, J.; Qian, X.; Yang, Y. Angew. Chem., Int. Ed. 2017, 56, 2979–2983. doi:10.1002/anie.201612301 |
| 9. | Staudinger, C.; Borisov, S. M. Methods Appl. Fluoresc. 2015, 3, 042005. doi:10.1088/2050-6120/3/4/042005 |
| 17. | Laursen, B. W.; Sørensen, T. J. J. Org. Chem. 2009, 74, 3183–3185. doi:10.1021/jo9002486 |
| 45. | Sørensen, T. J.; Kilså, K.; Laursen, B. W. Chem. – Eur. J. 2015, 21, 8521–8529. doi:10.1002/chem.201500355 |
| 46. | Sørensen, T. J.; Shi, D.; Laursen, B. W. Chem. – Eur. J. 2016, 22, 7046–7049. doi:10.1002/chem.201600496 |
| 6. | Moerner, W. E.; Orrit, M. Science 1999, 283, 1670–1676. doi:10.1126/science.283.5408.1670 |
| 7. | Grimm, J. B.; English, B. P.; Chen, J.; Slaughter, J. P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J. J.; Normanno, D.; Singer, R. H.; Lionnet, T.; Lavis, L. D. Nat. Methods 2015, 12, 244–250. doi:10.1038/nmeth.3256 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 29. | Wada, M.; Konishi, H.; Kirishima, K.; Takeuchi, H.; Natsume, S.; Erabi, T. Bull. Chem. Soc. Jpn. 1997, 70, 2737–2741. doi:10.1246/bcsj.70.2737 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
| 16. | Westerlund, F.; Hildebrandt, C. B.; Sørensen, T. J.; Laursen, B. W. Chem. – Eur. J. 2010, 16, 2992–2996. doi:10.1002/chem.200902965 |
| 22. | Laursen, B. W.; Reynisson, J.; Mikkelsen, K. V.; Bechgaard, K.; Harrit, N. Photochem. Photobiol. Sci. 2005, 4, 568–576. doi:10.1039/b501584g |
| 25. | Rich, R. M.; Stankowska, D. L.; Maliwal, B. P.; Sørensen, T. J.; Laursen, B. W.; Krishnamoorthy, R. R.; Gryczynski, Z.; Borejdo, J.; Gryczynski, I.; Fudala, R. Anal. Bioanal. Chem. 2013, 405, 2065–2075. doi:10.1007/s00216-012-6623-1 |
| 26. | Rich, R. M.; Mummert, M.; Gryczynski, Z.; Borejdo, J.; Sørensen, T. J.; Laursen, B. W.; Foldes-Papp, Z.; Gryczynski, I.; Fudala, R. Anal. Bioanal. Chem. 2013, 405, 4887–4894. doi:10.1007/s00216-013-6879-0 |
| 43. | Drexhage, K. H. Structure and Properties of Laser Dyes. In Dye Lasers; Schäfer, F. P., Ed.; Topics in Applied Physics, Vol. 1; Springer: Berlin, Heidelberg, Germany, 1973; pp 144–193. doi:10.1007/978-3-662-11579-4_4 |
| 44. | Griffiths, J. Colour and Constitution of Organic Molecules; Academic Press: London, 1976. |
| 19. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 20. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 21. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 13. | Meyer-Almes, F.-J. Methods Appl. Fluoresc. 2017, 5, 042002. doi:10.1088/2050-6120/aa7c7a |
| 27. | Hall, M. D.; Yasgar, A.; Peryea, T.; Braisted, J. C.; Jadhav, A.; Simeonov, A.; Coussens, N. P. Methods Appl. Fluoresc. 2016, 4, 022001. doi:10.1088/2050-6120/4/2/022001 |
| 28. | Sørensen, T. J.; Thyrhaug, E.; Szabelski, M.; Luchowski, R.; Gryczynski, I.; Gryczynski, Z.; Laursen, B. W. Methods Appl. Fluoresc. 2013, 1, 025001. doi:10.1088/2050-6120/1/2/025001 |
| 19. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 42. | Laleu, B.; Herse, C.; Laursen, B. W.; Bernardinelli, G.; Lacour, J. J. Org. Chem. 2003, 68, 6304–6308. doi:10.1021/jo0345998 |
| 16. | Westerlund, F.; Hildebrandt, C. B.; Sørensen, T. J.; Laursen, B. W. Chem. – Eur. J. 2010, 16, 2992–2996. doi:10.1002/chem.200902965 |
| 17. | Laursen, B. W.; Sørensen, T. J. J. Org. Chem. 2009, 74, 3183–3185. doi:10.1021/jo9002486 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
| 14. | Bosson, J.; Gouin, J.; Lacour, J. Chem. Soc. Rev. 2014, 43, 2824–2840. doi:10.1039/c3cs60461f |
| 15. | Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076 |
| 23. | Bogh, S. A.; Simmermacher, M.; Westberg, M.; Bregnhøj, M.; Rosenberg, M.; De Vico, L.; Veiga, M.; Laursen, B. W.; Ogilby, P. R.; Sauer, S. P. A.; Sørensen, T. J. ACS Omega 2017, 2, 193–203. doi:10.1021/acsomega.6b00211 |
| 24. | Bogh, S. A.; Bora, I.; Rosenberg, M.; Thyrhaug, E.; Laursen, B. W.; Sørensen, T. J. Methods Appl. Fluoresc. 2015, 3, 045001. doi:10.1088/2050-6120/3/4/045001 |
| 43. | Drexhage, K. H. Structure and Properties of Laser Dyes. In Dye Lasers; Schäfer, F. P., Ed.; Topics in Applied Physics, Vol. 1; Springer: Berlin, Heidelberg, Germany, 1973; pp 144–193. doi:10.1007/978-3-662-11579-4_4 |
| 44. | Griffiths, J. Colour and Constitution of Organic Molecules; Academic Press: London, 1976. |
| 32. | Martin, J. C.; Smith, R. G. J. Am. Chem. Soc. 1964, 86, 2252–2256. doi:10.1021/ja01065a030 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 32. | Martin, J. C.; Smith, R. G. J. Am. Chem. Soc. 1964, 86, 2252–2256. doi:10.1021/ja01065a030 |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
| 14. | Bosson, J.; Gouin, J.; Lacour, J. Chem. Soc. Rev. 2014, 43, 2824–2840. doi:10.1039/c3cs60461f |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 19. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 50. | Strickler, S. J.; Berg, R. A. J. Chem. Phys. 1962, 37, 814–822. doi:10.1063/1.1733166 |
| 40. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 41. | Brown, H. C.; Okamoto, Y. J. Am. Chem. Soc. 1958, 80, 4979–4987. doi:10.1021/ja01551a055 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
| 18. | Laursen, B. W.; Krebs, F. C.; Nielsen, M. F.; Bechgaard, K.; Christensen, J. B.; Harrit, N. J. Am. Chem. Soc. 1998, 120, 12255–12263. doi:10.1021/ja982550r |
| 39. | Simonsen, J. B.; Kjær, K.; Howes, P.; Nørgaard, K.; Bjørnholm, T.; Harrit, N.; Laursen, B. W. Langmuir 2009, 25, 3584–3592. doi:10.1021/la803733s |
| 37. | Rosenberg, M.; Rostgaard, K. R.; Liao, Z.; Madsen, A. Ø.; Martinez, K. L.; Vosch, T.; Laursen, B. W. Chem. Sci. 2018, 9, 3122–3130. doi:10.1039/c8sc00089a |
| 51. | Kotaskova, M.; Osman Oglou, O.; Helm, M. Org. Biomol. Chem. 2014, 12, 3816–3820. doi:10.1039/c4ob00533c |
| 38. | Rosenberg, M.; Santella, M.; Bogh, S. A.; Muñoz, A. V.; Andersen, H. O. B.; Hammerich, O.; Bora, I.; Lincke, K.; Laursen, B. W. J. Org. Chem. 2019, 84, 2556–2567. doi:10.1021/acs.joc.8b02978 |
| 33. | Laleu, B.; Mobian, P.; Herse, C.; Laursen, B. W.; Hopfgartner, G.; Bernardinelli, G.; Lacour, J. Angew. Chem., Int. Ed. 2005, 44, 1879–1883. doi:10.1002/anie.200462321 |
| 34. | Herse, C.; Bas, D.; Krebs, F. C.; Bürgi, T.; Weber, J.; Wesolowski, T.; Laursen, B. W.; Lacour, J. Angew. Chem., Int. Ed. 2003, 42, 3162–3166. doi:10.1002/anie.200351443 |
| 35. | Bosson, J.; Labrador, G. M.; Pascal, S.; Miannay, F.-A.; Yushchenko, O.; Li, H.; Bouffier, L.; Sojic, N.; Tovar, R. C.; Muller, G.; Jacquemin, D.; Laurent, A. D.; Le Guennic, B.; Vauthey, E.; Lacour, J. Chem. – Eur. J. 2016, 22, 18394–18403. doi:10.1002/chem.201603591 |
| 17. | Laursen, B. W.; Sørensen, T. J. J. Org. Chem. 2009, 74, 3183–3185. doi:10.1021/jo9002486 |
| 36. | Nicolas, C.; Bernardinelli, G.; Lacour, J. J. Phys. Org. Chem. 2010, 23, 1049–1056. doi:10.1002/poc.1753 |
| 31. | Sørensen, T. J.; Laursen, B. W. J. Org. Chem. 2010, 75, 6182–6190. doi:10.1021/jo1009917 |
© 2019 Santella et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)