Abstract
Sant-75 is a newly identified potent inhibitor of the hedgehog pathway. We designed a diversity-oriented synthesis program, and synthesized a series of Sant-75 analogues, which lays the foundation for further investigation of the structure–activity relationship of this important class of hedgehog-pathway inhibitors.

Graphical Abstract
Introduction
The Hedgehog (Hh) signaling pathway plays an essential role in embryonic development and adult tissue homeostasis in metazoans. The Hh ligands activate pathway signaling by binding to a 12-transmembrane protein receptor Patched (Ptch). In the unbound state, the Ptch receptor inhibits the activity of the downstream seven-pass transmembrane receptor Smoothened (Smo). Binding of Hh ligands to Ptch leads to the alleviation of this inhibition and eventually triggers activation of the glioma (Gli) family of transcription factors and their translocation to the nucleus. This activation results in the expression of specific genes that promote cell proliferation and differentiation [1,2]. The role of the Hh signaling pathway in human cancers was first confirmed through a study of Gorlin syndrome predisposing to basal cell carcinoma, arising from autosomal dominant mutations in Ptch [3]. Indeed, aberrant Hh signaling has been reported in a variety of other malignancy diseases, such as small-cell lung cancer, pancreatic cancer, prostate cancer, breast cancer and multiple myeloma [4-9]. Taken together, the development of Hh pathway antagonists has thus represented an attractive strategy for anticancer therapy [10,11]. Because mutated Ptch or Smo proteins are mostly responsible for the abnormal activation of Hh related to human diseases, intense efforts have been invested to identify therapeutic inhibitors acting on the Smo protein. Cyclopamine (Figure 1), a natural alkaloid isolated from Veratrum californicum [12,13], was disclosed as the first small molecule inhibitor of the Hh pathway through direct interaction with Smo [14,15]. Cyclopamine can effectively induce a decrease in proliferation and an increase of apoptosis in several murine models [16,17]. However, the clinical development of cyclopamine as a therapeutic in cancer is hampered by its poor aqueous solubility (ca. 5 µg/mL) and acid lability. Subsequently, Infinity Pharmaceuticals developed cyclopamine-based Smo inhibitors IPI-926 through structural modification on the A and D rings. IPI-926 exhibited improved pharmaceutical properties as well as a favorable pharmacokinetic profile, and showed complete tumor regression in a Hh-dependent medulloblastoma allograft model [18,19]. IPI-926 is currently being evaluated in the phase II trial of a safety and efficacy study of patients with metastatic or locally advanced (unresectable) chondrosarcoma and myelofibrosis [19,20]. Vismodegib (GDC-0449) [21-24], developed by Genentech and Curis, is another Smo antagonist which is progressing into the phase II clinical trial for the treatment of various cancers, including advanced basal cell carcinoma, and metastatic colorectal and ovarian cancers [25-27]. Recently, vismodegib was approved by the U.S. FDA to treat adult patients with basal cell carcinoma. In addition, a number of man-made inhibitors with a Smo binding affinity have been identified and reported [2,28-34], and some of them have entered phase I development.
![[1860-5397-8-94-1]](/bjoc/content/figures/1860-5397-8-94-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of Smo antagonists and agonists.
Figure 1: Structures of Smo antagonists and agonists.
SAG is a synthetic Hh pathway agonist that directly targets Smo in a manner that antagonizes cyclopamine action, and thus it may serve as an interesting scaffold for drug development [35,36]. Recently, we have identified a Smo antagonist Sant-75 through zebrafish-based screening of a SAG-derived chemical library [37]. Interestingly, this antagonist differs from agonist SAG only in the chain length of the secondary alkylamine due to the different conformational changes induced. This promising result prompted us to further investigate the structure–activity relationships (SAR) of Sant-75. Herein we describe our efforts in the development of synthetic methods for the construction of a library of Sant-75.
Results and Discussion
Chemistry
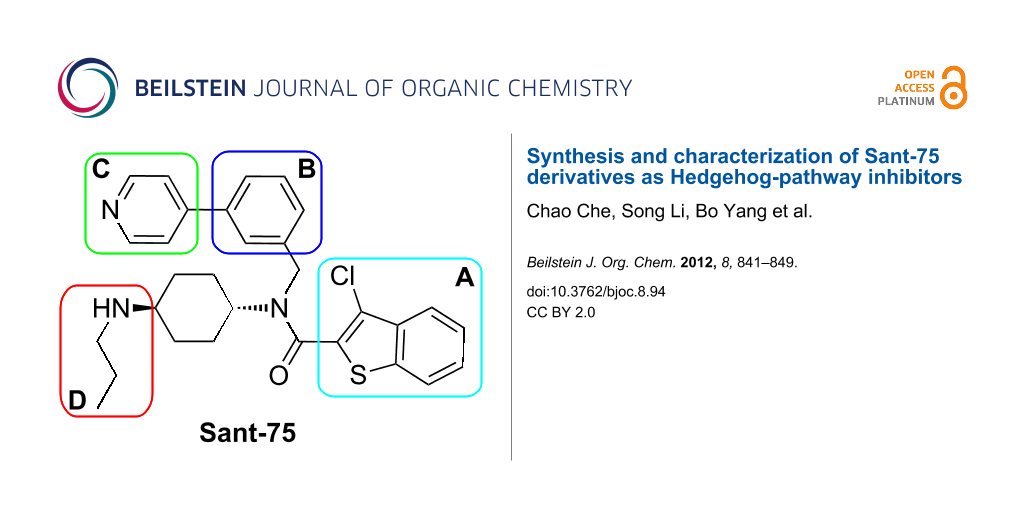
The scaffold of Sant-75 is divided into four distinct parts, namely 3-chlorobenzothiophene (motif A), a phenyl ring (motif B), 4-pyridine (motif C) and N-propyl-cyclohexane-1,4-diamine (motif D) (Scheme 1). In our earlier studies, the nature of the substituents on these regions was shown to have a profound effect on the activity. Examples of substituents that impart favorable activity include the alkyl group in region D.
![[1860-5397-8-94-i1]](/bjoc/content/inline/1860-5397-8-94-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General synthetic route for Sant-75. Reagents and conditions: (a) Pd(PPh3)4, PhMe, Na2CO3, H2O, 85 °C; (b) N-Boc-cyclohexane-1,4-diamine, THF, NaBH(OAc)3; (c) DMF, NaH, PrI, 0 °C to rt.; (d) 3-chlorobenzo[b]thiophene-2-carbonyl chloride, CH2Cl2, Et3N; (e) CH2Cl2, TFA.
Scheme 1: General synthetic route for Sant-75. Reagents and conditions: (a) Pd(PPh3)4, PhMe, Na2CO3, H2O, 85 ...
By modification of our first generation of synthetic methodology [38], the new general synthesis of the derivatives of Sant-75 is illustrated in Scheme 1. Accordingly, Suzuki coupling of 4-bromopyridine and 3-formylphenylboronic acid afforded biaryl aldehyde 2, which was then subjected to a reductive amination by condensation of aldehyde 2 with N-Boc-cyclohexane-1,4-diamine, followed by reduction with NaBH(OAc)3 to afford secondary amine 3. Selective alkylation of the newly generated secondary amine was achieved by treatment of amine 3 with NaH, followed by reaction with an alkylating reagent to give amine 4 in high yield. To complete the synthesis, amine 4 was reacted with acyl chloride in the presence of Et3N, and the formed amide was subjected to treatment with TFA to remove the Boc group.
Substituent-modifications on the motif A
The first series of derivatives is characterized by substituent modifications (Scheme 2, Scheme 3) and core modifications (Scheme 4) on the motif A. With respect to the substituent modifications, various groups, such as polar and hydrophobic groups, were introduced to the phenyl ring in motif A. Scheme 2 described the synthesis of derivatives 7a–l through the reaction of compound 4 with a number of substituted acyl chlorides 6a–l, which were prepared from the corresponding cinnamic acids by Higa cyclization [39,40]. It is noteworthy that some polar groups, including amino, hydroxy and sulfonamide, could not tolerate the conditions of Higa cyclization, and had to be introduced through transformation reactions after the N-acylation step.
![[1860-5397-8-94-i2]](/bjoc/content/inline/1860-5397-8-94-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substituent-modifications on the motif A. Reagents and conditions: (a) CH2Cl2, Et3N; (b) CH2Cl2, TFA.
Scheme 2: Substituent-modifications on the motif A. Reagents and conditions: (a) CH2Cl2, Et3N; (b) CH2Cl2, TF...
As depicted in Scheme 3, NH2-derivative 7m was prepared from the NO2-substituted precursor 7c by the reduction of the nitro group to an amino group using an FeCl3–Zn system [41]. Subsequently, capping the amino group with a sulfonyl or acetyl group led to 7n and 7o, respectively. In regard to hydroxy or acid modification, the demethylation or hydrolysis released the corresponding hydroxy group or carboxylic acid. Accordingly, 7e was treated with BBr3 in CH2Cl2 at −78 °C to afford the expected product 7p. 7d was directly subjected to basic hydrolysis in MeOH to yield carboxylic acid derivative 7q.
![[1860-5397-8-94-i3]](/bjoc/content/inline/1860-5397-8-94-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substituent-modifications on the motif A. Reagents and conditions: (a) (i) FeCl3, Zn, H2O, DMF, 100 °C; (ii) TFA, CH2Cl2; (b) (i) MeSO2Cl, CH2Cl2, Et3N; (ii) TFA, CH2Cl2; (c) (i) AcCl, CH2Cl2, Et3N; (ii) TFA, CH2Cl2; (d) BBr3, CH2Cl2, −78 °C; (e) KOH, MeOH, H2O.
Scheme 3: Substituent-modifications on the motif A. Reagents and conditions: (a) (i) FeCl3, Zn, H2O, DMF, 100...
To increase structural diversities, compound 4 was further reacted with various heteroaryl acids 8a–h to yield 9a–h (Scheme 4). The following considerations were taken for the selection of the heteroaryl acids: removal of the Cl atom (8a); replacement of the S atom with N or O atoms (8b, 8c); two heteroatoms in a five-membered ring (8d–f); and a 5,5-fused ring heterocycle (8h), all of which will help us to more distinctly study the SAR in motif A. Compounds 8a–h were either synthesized using reported methods [42-46] or are commercially available.
![[1860-5397-8-94-i4]](/bjoc/content/inline/1860-5397-8-94-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Core modification on the motif A. Reagents and conditions: (a) BOP, DIEA, DMF; (b) CH2Cl2, TFA.
Scheme 4: Core modification on the motif A. Reagents and conditions: (a) BOP, DIEA, DMF; (b) CH2Cl2, TFA.
Substituent-modifications on the motif B
In the second series of novel scaffolds, structural modification was focused on alternatives to the phenyl ring to improve aqueous solubility. As illustrated in Figure 2, we replaced the phenyl ring with a variety of structurally diverse heteroaryl groups, including pyridine, pyrimidine and imidazole rings. In order to investigate the effect of the N-position in the heteroaryl ring, different pyridine and pyrimidine substituents were also incorporated. Generally, the compounds 10a–g were synthesized following the general route in Scheme 1, by using aldehydes 11a–g as key intermediates. As described in Scheme 5, 11a–d were prepared through Pd-catalyzed Suzuki coupling of 4-pyridylboronic acid with corresponding formyl-functionalized pyridylbromides 12a–d, which were commercially available. However, application of the reaction conditions identical to those used in the synthesis of compound 2 (Scheme 1) resulted in very low yields. After systematic optimization of the reaction conditions, we found that the reaction proceeded well in 1,4-dioxane at 100 °C for 8 h with Na2CO3 as base in the presence of Pd(OAc)2 as catalyst, and PPh3 as ligand, providing the products in 73–85% yield.
![[1860-5397-8-94-2]](/bjoc/content/figures/1860-5397-8-94-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Core modification on the motif B.
Figure 2: Core modification on the motif B.
![[1860-5397-8-94-i5]](/bjoc/content/inline/1860-5397-8-94-i5.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of key intermediate biaryl aldehydes. Reagents and conditions: (a) Pd(OAc)2, PPh3, 1,4-dioxane, Na2CO3, 100 °C; (b) cat. NaOMe, MeOH; then NH4Cl; (c) acetylacetaldehyde dimethyl acetal, 1,4-dioxane, reflux; (d) SeO2, 1,4-dioxane, reflux; (e) DMF-DMA, PhMe, reflux; (f) acetamidine hydrochloride, Na, EtOH, reflux; (g) 1H-imidazole-4-carbaldehyde, DMF, CuI, Cs2CO3.
Scheme 5: Synthesis of key intermediate biaryl aldehydes. Reagents and conditions: (a) Pd(OAc)2, PPh3, 1,4-di...
Different from the synthetic strategy of 11a–d, the pyrimidine nucleus in biaryl aldehydes 11e–f had to be constructed through condensation of a protected β-ketoaldehyde with the corresponding amidine. Compound 11e was synthesized from isonicotinonitrile through three steps described previously. Isonicotinonitrile was converted to isonicotinamidine hydrochloride 13 upon reaction in MeOH in the presence of catalytic NaOMe, followed by treatment with NH4Cl. Then, condensation of 13 with acetylacetaldehyde dimethyl acetal in 1,4-dioxane afforded 6-methyl-2-(4-pyridinyl)pyrimidine 14 in 62% yield [47]. Consequent allylic oxidation of 14 with selenium dioxide gave 11e in moderate yield. Likewise, compound 11f was prepared though SeO2-oxidation of 16, which was prepared by the reported method using 4-acetylpyridine as the starting material [48]. 4-Acetylpyridine was converted to enaminone intermediates 15 upon treatment with DMF-DMA in toluene under reflux, followed by condensation cyclization with acetamidine to give 16. Compound 11g could be installed in single step by Ullmann coupling of 17 with 4-bromopyridine, in good yield.
Substituent-modifications on the motif C
In the third round of structural modifications, chemical diversity was explored by varying the nature of the heterocycles (motif C) and the position of the nitrogen atom in pyridine. As shown in Scheme 6, para-substituted pyridine was first replaced with meta- and ortho-pyridine, and other heterocycles, including pyrimidines and five-membered heteroaryl rings, as well as nonaromatic heterocycles, such as morpholine, piperazine and homo-piperazine, were also investigated. Accordingly, a small series of Sant-75 analogues 18a–j were synthesized in a parallel fashion from the corresponding intermediates 19a–j. For intermediates 19a–j, different coupling reactions were required to achieve their synthesis, depending on the type of structure. 19a–d were prepared through Suzuki coupling as described above, and 19e–g were obtained in 70–85% yield by Ullmann coupling of 3-bromobenzaldehyde with an excess of the appropriate amines 20e–g [49]. The Buchwald coupling of protected 3-bromobenzaldehyde with amines 20h–j proceeded smoothly in toluene with NaOt-Bu as base by using a Pd2(dba)3/rac-BINAP catalysis system [50].
![[1860-5397-8-94-i6]](/bjoc/content/inline/1860-5397-8-94-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Chemical modifications on the motif C. Reagents and conditions: (a) Pd(OAc)2, PPh3, 1,4-dioxane, Na2CO3, 100 °C; (b) CuI, DMF, Cs2CO3, 120 °C; (c) (i) Pd2(dba)3, rac-BINAP, NaOt-Bu, PhMe, 100 °C; (ii) aq. HCl, THF.
Scheme 6: Chemical modifications on the motif C. Reagents and conditions: (a) Pd(OAc)2, PPh3, 1,4-dioxane, Na2...
Substituent-modifications on the motif D
It has previously been shown that the substitution pattern on the nitrogen atom of trans-1,4-diaminocyclohexane moiety plays a critical role in the activity of the compounds as Hedgehog-pathway inhibitors. Therefore, the emphasis of the fourth series of structural optimization was placed on the nitrogen atom through capping of the amino group with various acyl chlorides, delivering the corresponding amides 21a–j. It should be noted that the capping of the amino group can increase the metabolic stability of the compounds. The R2 group in modified compounds was extensively studied, including alkyl, cycloalkyl, phenyl and heteroaryl groups, and a selection of the generated compounds is listed in Scheme 7.
![[1860-5397-8-94-i7]](/bjoc/content/inline/1860-5397-8-94-i7.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Chemical modifications on the motif D. Reagents and conditions: (a) R2COCl, CH2Cl2.
Scheme 7: Chemical modifications on the motif D. Reagents and conditions: (a) R2COCl, CH2Cl2.
Conclusion
By modification of our first generation of synthesis of SAG derivatives, a second generation of the synthetic program was established, which allows structurally diverse derivatives of Sant-75 to be synthesized in a systematic manner. The constructed library was fully characterized, which provides a foundation for the further investigation of their biological activities. The biological investigation of this library is currently underway in our laboratories, and the results will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 268.1 KB | Download |
Acknowledgements
This project was supported by the National Science and Technology Major Project entitled the “Development of Key Technology for the Combinatorial Synthesis of Privileged Scaffolds” (2009ZX09501-012), the National Basic Research Program of China (973 Program, Grant No. 2010CB833201), the National Natural Science Foundation of China (Grant No. 21102006 to C.C.), the Natural Science Foundation of Guangdong Province (Grant No. S2011010000185 to C.C.), and the Shenzhen Biological Industry Development Foundation (Grant No. JSA201005310118A to L.S.).
References
-
Rohatgi, R.; Scott, M. P. Nat. Cell Biol. 2007, 9, 1005–1009. doi:10.1038/ncb435
Return to citation in text: [1] -
Peukert, S.; Miller-Moslin, K. ChemMedChem 2010, 5, 500–512. doi:10.1002/cmdc.201000011
Return to citation in text: [1] [2] -
Johnson, R. L.; Rothman, A. L.; Xie, J.; Goodrich, L. V.; Bare, J. W.; Bonifas, J. M.; Quinn, A. G.; Myers, R. M.; Cox, D. R.; Epstein, E. H., Jr.; Scott, M. P. Science 1996, 272, 1668–1671. doi:10.1126/science.272.5268.1668
Return to citation in text: [1] -
Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Nat. Rev. Cancer 2002, 2, 361–372. doi:10.1038/nrc796
Return to citation in text: [1] -
Watkins, D. N.; Berman, D. M.; Burkholder, S. G.; Wang, B.; Beachy, P. A.; Baylin, S. B. Nature 2003, 422, 313–317. doi:10.1038/nature01493
Return to citation in text: [1] -
Yauch, R. L.; Gould, S. E.; Scales, S. J.; Tang, T.; Tian, H.; Ahn, C. P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; Nannini-Pepe, M.; Kotkow, K.; Marsters, J. C.; Rubin, L. L.; de Sauvage, F. J. Nature 2008, 455, 406–410. doi:10.1038/nature07275
Return to citation in text: [1] -
Karhadkar, S. S.; Bova, G. S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J. T.; Berman, D. M.; Beachy, P. A. Nature 2004, 431, 707–712. doi:10.1038/nature02962
Return to citation in text: [1] -
Liu, S.; Dontu, G.; Mantle, I. D.; Patel, S.; Ahn, N.; Jackson, K. W.; Suri, P.; Wicha, M. S. Cancer Res. 2006, 66, 6063–6071. doi:10.1158/0008-5472.CAN-06-0054
Return to citation in text: [1] -
Peacock, C. D.; Wang, Q.; Gesell, G. S.; Corcoran-Schwartz, I. M.; Jones, E.; Kim, J.; Devereux, W. L.; Rhodes, J. T.; Huff, C. A.; Beachy, P. A.; Watkins, D. N.; Matsui, W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4048–4053. doi:10.1073/pnas.0611682104
Return to citation in text: [1] -
Rubin, L. L.; de Sauvage, F. J. Nat. Rev. Drug Discovery 2006, 5, 1026–1033. doi:10.1038/nrd2086
Return to citation in text: [1] -
Borzillo, G. V.; Lippa, B. Curr. Top. Med. Chem. 2005, 5, 147–157. doi:10.2174/1568026053507732
Return to citation in text: [1] -
Keller, R. F. Phytochemistry 1968, 7, 303–306. doi:10.1016/S0031-9422(00)86328-1
Return to citation in text: [1] -
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem., Int. Ed. 2010, 49, 3418–3427. doi:10.1002/anie.200906967
Return to citation in text: [1] -
Cooper, M. K.; Porter, J. A.; Young, K. E.; Beachy, P. A. Science 1998, 280, 1603–1607. doi:10.1126/science.280.5369.1603
Return to citation in text: [1] -
Chen, J. K.; Taipale, J.; Cooper, M. K.; Beachy, P. A. Genes Dev. 2002, 16, 2743–2748. doi:10.1101/gad.1025302
Return to citation in text: [1] -
Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M. R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; Warmuth, M. Cancer Cell 2008, 14, 238–249. doi:10.1016/j.ccr.2008.08.003
Return to citation in text: [1] -
Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944–951. doi:10.1038/nm1614
Return to citation in text: [1] -
Tremblay, M. R.; Nevalainen, M.; Nair, S. J.; Porter, J. R.; Castro, A. C.; Behnke, M. L.; Yu, L.-C.; Hagel, M.; White, K.; Faia, K.; Grenier, L.; Campbell, M. J.; Cushing, J.; Woodward, C. N.; Hoyt, J.; Foley, M. A.; Read, M. A.; Sydor, J. R.; Tong, J. K.; Palombella, V. J.; McGovern, K.; Adams, J. J. Med. Chem. 2008, 51, 6646–6649. doi:10.1021/jm8008508
Return to citation in text: [1] -
Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.; Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.; Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.; Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400–4418. doi:10.1021/jm900305z
Return to citation in text: [1] [2] -
A phase 2 study with IPI-926 in patients with myelofibrosis. http://www.clinicaltrials.gov/ct2/show/NCT01371617?term=IPI+926+myelofibrosis&rank=1 (accessed Feb 27, 2012).
Return to citation in text: [1] -
Robarge, K. D.; Brunton, S. A.; Castanedo, G. M.; Cui, Y.; Dina, M. S.; Goldsmith, R.; Gould, S. E.; Guichert, O.; Gunzner, J. L.; Halladay, J.; Jia, W.; Khojasteh, C.; Koehler, M. F. T.; Kotkow, K.; La, H.; LaLonde, R. L.; Lau, K.; Lee, L.; Marshall, D.; Marsters, J. C., Jr.; Murray, L. J.; Qian, C.; Rubin, L. L.; Salphati, L.; Stanley, M. S.; Stibbard, J. H. A.; Sutherlin, D. P.; Ubhayaker, S.; Wang, S.; Wong, S.; Xie, M. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. doi:10.1016/j.bmcl.2009.08.049
Return to citation in text: [1] -
Giannetti, A. M.; Wong, H.; Dijkgraaf, G. J. P.; Dueber, E. C.; Ortwine, D. F.; Bravo, B. J.; Gould, S. E.; Plise, E. G.; Lum, B. L.; Malhi, V.; Graham, R. A. J. Med. Chem. 2011, 54, 2592–2601. doi:10.1021/jm1008924
Return to citation in text: [1] -
Rudin, C. M.; Hann, C. L.; Laterra, J.; Yauch, R. L.; Callahan, C. A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S. E.; Coleman, B.; LoRusso, P. M.; Von Hoff, D. D.; de Sauvage, F. J.; Low, J. A. N. Engl. J. Med. 2009, 361, 1173–1178. doi:10.1056/NEJMoa0902903
Return to citation in text: [1] -
Von Hoff, D. D.; LoRusso, P. M.; Rudin, C. M.; Reddy, J. C.; Yauch, R. L.; Tibes, R.; Weiss, G. J.; Borad, M. J.; Hann, C. L.; Brahmer, J. R.; Mackey, H. M.; Lum, B. L.; Darbonne, W. C.; Marsters, J. C., Jr.; de Sauvage, F. J.; Low, J. A. N. Engl. J. Med. 2009, 361, 1164–1172. doi:10.1056/NEJMoa0905360
Return to citation in text: [1] -
A study evaluating the efficacy and safety of GDC-0449 (Hedgehog pathway inhibitor) in patients with advanced basal cell carcinoma. http://clinicaltrials.gov/ct2/show/NCT00833417?term=GDC-0449&rank=1 (accessed Feb 27, 2012).
Return to citation in text: [1] -
A study of GDC-0449 (Hedgehog pathway inhibitor) with concurrent chemotherapy and bevacizumab as first-line therapy for metastatic colorectal cancer. http://clinicaltrials.gov/ct2/show/NCT00636610?term=GDC-0449&rank=4 (accessed Feb 27, 2012).
Return to citation in text: [1] -
A study of GDC-0449 (Hedgehog pathway inhibitor) as maintenance therapy in patients with ovarian cancer in a second or third complete remission, http://clinicaltrials.gov/ct2/show/NCT00739661?term=GDC-0449&rank=5 (accessed Feb 27, 2012).
Return to citation in text: [1] -
Mahindroo, N.; Punchihewa, C.; Fujii, N. J. Med. Chem. 2009, 52, 3829–3845. doi:10.1021/jm801420y
Return to citation in text: [1] -
Miller-Moslin, K.; Peukert, S.; Jain, R. K.; McEwan, M. A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; Dai, M.; Perez, L.; Michael, W.; Sheng, T.; Lei, H.; Zhang, R.; Williams, J.; Bourret, A.; Ramamurthy, A.; Yuan, J.; Guo, R.; Matsumoto, M.; Vattay, A.; Maniara, W.; Amaral, A.; Dorsch, M.; Kelleher, J. F., III. J. Med. Chem. 2009, 52, 3954–3968. doi:10.1021/jm900309j
Return to citation in text: [1] -
Bahceci, S.; Bajjalieh, W.; Chen, J.; Epshteyn, S.; Forsyth, T. P.; Huynh, T. P.; Kim, B. G.; Leahy, J. W.; Lee, M. S.; Lewis, G. L.; Mac, M. B.; Mann, G.; Marlowe, C. K.; Ridgway, B. H.; Sangalang, J. C.; Shi, X.; Takeuchi, C. S.; Wang, Y. Inhibitors of the hedgehog pathway. WO Patent 2008112913, Sept 18, 2008.
Return to citation in text: [1] -
Munchhof, M. J.; Reiter, L. A.; Shavnya, A.; Jones, C. S.; Li, Q.; Linde, R. G., II. Benzimidazole derivatives. WO Patent 2008075196, June 26, 2008.
Return to citation in text: [1] -
Peukert, S.; Jain, R. K.; Geisser, A.; Sun, Y.; Zhang, R.; Bourret, A.; Carlson, A.; DaSilva, J.; Ramamurthy, A.; Kelleher, J. F. Bioorg. Med. Chem. Lett. 2009, 19, 328–331. doi:10.1016/j.bmcl.2008.11.096
Return to citation in text: [1] -
Büttner, A.; Seifert, K.; Cottin, T.; Sarli, V.; Tzagkaroulaki, L.; Scholz, S.; Giannis, A. Bioorg. Med. Chem. 2009, 17, 4943–4954. doi:10.1016/j.bmc.2009.06.008
Return to citation in text: [1] -
Stanton, B. Z.; Peng, L. F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J. L.; Taveras, K. M.; Hyman, J. M.; Lee, S. W.; Koehler, A. N.; Chen, J. K.; Fox, J. L.; Mandinova, A.; Schreiber, S. L. Nat. Chem. Biol. 2009, 5, 154–156. doi:10.1038/nchembio.142
Return to citation in text: [1] -
Frank-Kamenetsky, M.; Zhang, X. M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F. Y.; Jones, S.; Shulok, J.; Rubin, L. L.; Porter, J. A. J. Biol. 2002, 1, No. 10. doi:10.1186/1475-4924-1-10
Return to citation in text: [1] -
Chen, J. K.; Taipale, J.; Young, K. E.; Maiti, T.; Beachy, P. A. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14071–14076. doi:10.1073/pnas.182542899
Return to citation in text: [1] -
Yang, H.; Xiang, J.; Wang, N.; Zhao, Y.; Hyman, J.; Li, S.; Jiang, J.; Chen, J. K.; Yang, Z.; Lin, S. J. Biol. Chem. 2009, 284, 20876–20884. doi:10.1074/jbc.M807648200
Return to citation in text: [1] -
Wang, N.; Xiang, J.; Ma, Z.; Quan, J.; Chen, J.; Yang, Z. J. Comb. Chem. 2008, 10, 825–834. doi:10.1021/cc800025n
Return to citation in text: [1] -
Higa, T.; Krubsack, A. J. J. Org. Chem. 1975, 40, 3037–3045. doi:10.1021/jo00909a007
Return to citation in text: [1] -
Higa, T.; Krubsack, A. J. J. Org. Chem. 1976, 41, 3399–3403. doi:10.1021/jo00883a014
Return to citation in text: [1] -
Desai, D. G.; Swami, S. S.; Hapase, S. B. Synth. Commun. 1999, 29, 1033–1036. doi:10.1080/00397919908086068
Return to citation in text: [1] -
Cole, D. C.; Du, X.; Hotchandani, R.; Hu, Y.; Levin, J. I.; Li, J.; Li, W.; Lovering, F.; Rush, T. S., III; Tam, S. Y.; Wu, J.; Xiang, J. S. Biaryl sulfonamides and methods for using same. US Patent US020050143422, June 30, 2005.
Return to citation in text: [1] -
Campaigne, E.; Van Verth, J. E. J. Org. Chem. 1958, 23, 1344–1346. doi:10.1021/jo01103a029
Return to citation in text: [1] -
Thakurdesai, P. A.; Wadodkar, S. G.; Chopade, C. T. Pharmacologyonline 2007, 1, 314–329.
Return to citation in text: [1] -
Wang, Z.; Vince, R. Bioorg. Med. Chem. 2008, 16, 3587–3595. doi:10.1016/j.bmc.2008.02.007
Return to citation in text: [1] -
Zhu, G.-D.; Gunawardana, I. W.; Boyd, S. A.; Melcher, L. M. J. Heterocycl. Chem. 2008, 45, 91–96. doi:10.1002/jhet.5570450106
Return to citation in text: [1] -
Kamei, K.; Maeda, N.; Katsuragi-Ogino, R.; Koyama, M.; Nakajima, M.; Tatsuoka, T.; Ohno, T.; Inoue, T. Bioorg. Med. Chem. Lett. 2005, 15, 2990–2993. doi:10.1016/j.bmcl.2005.04.059
Return to citation in text: [1] -
Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B., Jr. J. Med. Chem. 1978, 21, 623–628. doi:10.1021/jm00205a006
Return to citation in text: [1] -
Zhu, L.; Guo, P.; Li, G.; Lan, J.; Xie, R.; You, J. J. Org. Chem. 2007, 72, 8535–8538. doi:10.1021/jo0712289
Return to citation in text: [1] -
Jean, L.; Rouden, J.; Maddaluno, J.; Lasne, M.-C. J. Org. Chem. 2004, 69, 8893–8902. doi:10.1021/jo0487193
Return to citation in text: [1]
| 47. | Kamei, K.; Maeda, N.; Katsuragi-Ogino, R.; Koyama, M.; Nakajima, M.; Tatsuoka, T.; Ohno, T.; Inoue, T. Bioorg. Med. Chem. Lett. 2005, 15, 2990–2993. doi:10.1016/j.bmcl.2005.04.059 |
| 41. | Desai, D. G.; Swami, S. S.; Hapase, S. B. Synth. Commun. 1999, 29, 1033–1036. doi:10.1080/00397919908086068 |
| 42. | Cole, D. C.; Du, X.; Hotchandani, R.; Hu, Y.; Levin, J. I.; Li, J.; Li, W.; Lovering, F.; Rush, T. S., III; Tam, S. Y.; Wu, J.; Xiang, J. S. Biaryl sulfonamides and methods for using same. US Patent US020050143422, June 30, 2005. |
| 43. | Campaigne, E.; Van Verth, J. E. J. Org. Chem. 1958, 23, 1344–1346. doi:10.1021/jo01103a029 |
| 44. | Thakurdesai, P. A.; Wadodkar, S. G.; Chopade, C. T. Pharmacologyonline 2007, 1, 314–329. |
| 45. | Wang, Z.; Vince, R. Bioorg. Med. Chem. 2008, 16, 3587–3595. doi:10.1016/j.bmc.2008.02.007 |
| 46. | Zhu, G.-D.; Gunawardana, I. W.; Boyd, S. A.; Melcher, L. M. J. Heterocycl. Chem. 2008, 45, 91–96. doi:10.1002/jhet.5570450106 |
| 1. | Rohatgi, R.; Scott, M. P. Nat. Cell Biol. 2007, 9, 1005–1009. doi:10.1038/ncb435 |
| 2. | Peukert, S.; Miller-Moslin, K. ChemMedChem 2010, 5, 500–512. doi:10.1002/cmdc.201000011 |
| 12. | Keller, R. F. Phytochemistry 1968, 7, 303–306. doi:10.1016/S0031-9422(00)86328-1 |
| 13. | Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem., Int. Ed. 2010, 49, 3418–3427. doi:10.1002/anie.200906967 |
| 38. | Wang, N.; Xiang, J.; Ma, Z.; Quan, J.; Chen, J.; Yang, Z. J. Comb. Chem. 2008, 10, 825–834. doi:10.1021/cc800025n |
| 10. | Rubin, L. L.; de Sauvage, F. J. Nat. Rev. Drug Discovery 2006, 5, 1026–1033. doi:10.1038/nrd2086 |
| 11. | Borzillo, G. V.; Lippa, B. Curr. Top. Med. Chem. 2005, 5, 147–157. doi:10.2174/1568026053507732 |
| 39. | Higa, T.; Krubsack, A. J. J. Org. Chem. 1975, 40, 3037–3045. doi:10.1021/jo00909a007 |
| 40. | Higa, T.; Krubsack, A. J. J. Org. Chem. 1976, 41, 3399–3403. doi:10.1021/jo00883a014 |
| 4. | Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Nat. Rev. Cancer 2002, 2, 361–372. doi:10.1038/nrc796 |
| 5. | Watkins, D. N.; Berman, D. M.; Burkholder, S. G.; Wang, B.; Beachy, P. A.; Baylin, S. B. Nature 2003, 422, 313–317. doi:10.1038/nature01493 |
| 6. | Yauch, R. L.; Gould, S. E.; Scales, S. J.; Tang, T.; Tian, H.; Ahn, C. P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; Nannini-Pepe, M.; Kotkow, K.; Marsters, J. C.; Rubin, L. L.; de Sauvage, F. J. Nature 2008, 455, 406–410. doi:10.1038/nature07275 |
| 7. | Karhadkar, S. S.; Bova, G. S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J. T.; Berman, D. M.; Beachy, P. A. Nature 2004, 431, 707–712. doi:10.1038/nature02962 |
| 8. | Liu, S.; Dontu, G.; Mantle, I. D.; Patel, S.; Ahn, N.; Jackson, K. W.; Suri, P.; Wicha, M. S. Cancer Res. 2006, 66, 6063–6071. doi:10.1158/0008-5472.CAN-06-0054 |
| 9. | Peacock, C. D.; Wang, Q.; Gesell, G. S.; Corcoran-Schwartz, I. M.; Jones, E.; Kim, J.; Devereux, W. L.; Rhodes, J. T.; Huff, C. A.; Beachy, P. A.; Watkins, D. N.; Matsui, W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4048–4053. doi:10.1073/pnas.0611682104 |
| 35. | Frank-Kamenetsky, M.; Zhang, X. M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F. Y.; Jones, S.; Shulok, J.; Rubin, L. L.; Porter, J. A. J. Biol. 2002, 1, No. 10. doi:10.1186/1475-4924-1-10 |
| 36. | Chen, J. K.; Taipale, J.; Young, K. E.; Maiti, T.; Beachy, P. A. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14071–14076. doi:10.1073/pnas.182542899 |
| 3. | Johnson, R. L.; Rothman, A. L.; Xie, J.; Goodrich, L. V.; Bare, J. W.; Bonifas, J. M.; Quinn, A. G.; Myers, R. M.; Cox, D. R.; Epstein, E. H., Jr.; Scott, M. P. Science 1996, 272, 1668–1671. doi:10.1126/science.272.5268.1668 |
| 37. | Yang, H.; Xiang, J.; Wang, N.; Zhao, Y.; Hyman, J.; Li, S.; Jiang, J.; Chen, J. K.; Yang, Z.; Lin, S. J. Biol. Chem. 2009, 284, 20876–20884. doi:10.1074/jbc.M807648200 |
| 19. | Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.; Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.; Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.; Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400–4418. doi:10.1021/jm900305z |
| 20. | A phase 2 study with IPI-926 in patients with myelofibrosis. http://www.clinicaltrials.gov/ct2/show/NCT01371617?term=IPI+926+myelofibrosis&rank=1 (accessed Feb 27, 2012). |
| 25. | A study evaluating the efficacy and safety of GDC-0449 (Hedgehog pathway inhibitor) in patients with advanced basal cell carcinoma. http://clinicaltrials.gov/ct2/show/NCT00833417?term=GDC-0449&rank=1 (accessed Feb 27, 2012). |
| 26. | A study of GDC-0449 (Hedgehog pathway inhibitor) with concurrent chemotherapy and bevacizumab as first-line therapy for metastatic colorectal cancer. http://clinicaltrials.gov/ct2/show/NCT00636610?term=GDC-0449&rank=4 (accessed Feb 27, 2012). |
| 27. | A study of GDC-0449 (Hedgehog pathway inhibitor) as maintenance therapy in patients with ovarian cancer in a second or third complete remission, http://clinicaltrials.gov/ct2/show/NCT00739661?term=GDC-0449&rank=5 (accessed Feb 27, 2012). |
| 50. | Jean, L.; Rouden, J.; Maddaluno, J.; Lasne, M.-C. J. Org. Chem. 2004, 69, 8893–8902. doi:10.1021/jo0487193 |
| 18. | Tremblay, M. R.; Nevalainen, M.; Nair, S. J.; Porter, J. R.; Castro, A. C.; Behnke, M. L.; Yu, L.-C.; Hagel, M.; White, K.; Faia, K.; Grenier, L.; Campbell, M. J.; Cushing, J.; Woodward, C. N.; Hoyt, J.; Foley, M. A.; Read, M. A.; Sydor, J. R.; Tong, J. K.; Palombella, V. J.; McGovern, K.; Adams, J. J. Med. Chem. 2008, 51, 6646–6649. doi:10.1021/jm8008508 |
| 19. | Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.; Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.; Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.; Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400–4418. doi:10.1021/jm900305z |
| 2. | Peukert, S.; Miller-Moslin, K. ChemMedChem 2010, 5, 500–512. doi:10.1002/cmdc.201000011 |
| 28. | Mahindroo, N.; Punchihewa, C.; Fujii, N. J. Med. Chem. 2009, 52, 3829–3845. doi:10.1021/jm801420y |
| 29. | Miller-Moslin, K.; Peukert, S.; Jain, R. K.; McEwan, M. A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; Dai, M.; Perez, L.; Michael, W.; Sheng, T.; Lei, H.; Zhang, R.; Williams, J.; Bourret, A.; Ramamurthy, A.; Yuan, J.; Guo, R.; Matsumoto, M.; Vattay, A.; Maniara, W.; Amaral, A.; Dorsch, M.; Kelleher, J. F., III. J. Med. Chem. 2009, 52, 3954–3968. doi:10.1021/jm900309j |
| 30. | Bahceci, S.; Bajjalieh, W.; Chen, J.; Epshteyn, S.; Forsyth, T. P.; Huynh, T. P.; Kim, B. G.; Leahy, J. W.; Lee, M. S.; Lewis, G. L.; Mac, M. B.; Mann, G.; Marlowe, C. K.; Ridgway, B. H.; Sangalang, J. C.; Shi, X.; Takeuchi, C. S.; Wang, Y. Inhibitors of the hedgehog pathway. WO Patent 2008112913, Sept 18, 2008. |
| 31. | Munchhof, M. J.; Reiter, L. A.; Shavnya, A.; Jones, C. S.; Li, Q.; Linde, R. G., II. Benzimidazole derivatives. WO Patent 2008075196, June 26, 2008. |
| 32. | Peukert, S.; Jain, R. K.; Geisser, A.; Sun, Y.; Zhang, R.; Bourret, A.; Carlson, A.; DaSilva, J.; Ramamurthy, A.; Kelleher, J. F. Bioorg. Med. Chem. Lett. 2009, 19, 328–331. doi:10.1016/j.bmcl.2008.11.096 |
| 33. | Büttner, A.; Seifert, K.; Cottin, T.; Sarli, V.; Tzagkaroulaki, L.; Scholz, S.; Giannis, A. Bioorg. Med. Chem. 2009, 17, 4943–4954. doi:10.1016/j.bmc.2009.06.008 |
| 34. | Stanton, B. Z.; Peng, L. F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J. L.; Taveras, K. M.; Hyman, J. M.; Lee, S. W.; Koehler, A. N.; Chen, J. K.; Fox, J. L.; Mandinova, A.; Schreiber, S. L. Nat. Chem. Biol. 2009, 5, 154–156. doi:10.1038/nchembio.142 |
| 16. | Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M. R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; Warmuth, M. Cancer Cell 2008, 14, 238–249. doi:10.1016/j.ccr.2008.08.003 |
| 17. | Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944–951. doi:10.1038/nm1614 |
| 48. | Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B., Jr. J. Med. Chem. 1978, 21, 623–628. doi:10.1021/jm00205a006 |
| 14. | Cooper, M. K.; Porter, J. A.; Young, K. E.; Beachy, P. A. Science 1998, 280, 1603–1607. doi:10.1126/science.280.5369.1603 |
| 15. | Chen, J. K.; Taipale, J.; Cooper, M. K.; Beachy, P. A. Genes Dev. 2002, 16, 2743–2748. doi:10.1101/gad.1025302 |
| 21. | Robarge, K. D.; Brunton, S. A.; Castanedo, G. M.; Cui, Y.; Dina, M. S.; Goldsmith, R.; Gould, S. E.; Guichert, O.; Gunzner, J. L.; Halladay, J.; Jia, W.; Khojasteh, C.; Koehler, M. F. T.; Kotkow, K.; La, H.; LaLonde, R. L.; Lau, K.; Lee, L.; Marshall, D.; Marsters, J. C., Jr.; Murray, L. J.; Qian, C.; Rubin, L. L.; Salphati, L.; Stanley, M. S.; Stibbard, J. H. A.; Sutherlin, D. P.; Ubhayaker, S.; Wang, S.; Wong, S.; Xie, M. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. doi:10.1016/j.bmcl.2009.08.049 |
| 22. | Giannetti, A. M.; Wong, H.; Dijkgraaf, G. J. P.; Dueber, E. C.; Ortwine, D. F.; Bravo, B. J.; Gould, S. E.; Plise, E. G.; Lum, B. L.; Malhi, V.; Graham, R. A. J. Med. Chem. 2011, 54, 2592–2601. doi:10.1021/jm1008924 |
| 23. | Rudin, C. M.; Hann, C. L.; Laterra, J.; Yauch, R. L.; Callahan, C. A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S. E.; Coleman, B.; LoRusso, P. M.; Von Hoff, D. D.; de Sauvage, F. J.; Low, J. A. N. Engl. J. Med. 2009, 361, 1173–1178. doi:10.1056/NEJMoa0902903 |
| 24. | Von Hoff, D. D.; LoRusso, P. M.; Rudin, C. M.; Reddy, J. C.; Yauch, R. L.; Tibes, R.; Weiss, G. J.; Borad, M. J.; Hann, C. L.; Brahmer, J. R.; Mackey, H. M.; Lum, B. L.; Darbonne, W. C.; Marsters, J. C., Jr.; de Sauvage, F. J.; Low, J. A. N. Engl. J. Med. 2009, 361, 1164–1172. doi:10.1056/NEJMoa0905360 |
| 49. | Zhu, L.; Guo, P.; Li, G.; Lan, J.; Xie, R.; You, J. J. Org. Chem. 2007, 72, 8535–8538. doi:10.1021/jo0712289 |
© 2012 Che et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)