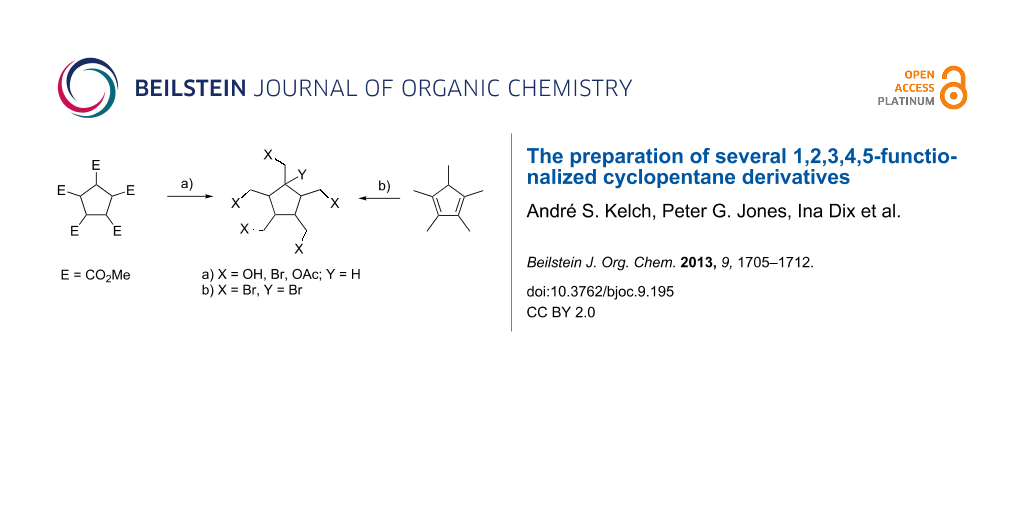
Abstract
With the goal of eventually synthesizing [5]radialene (3), the still missing member of the parent radialene hydrocarbons, we have prepared the pentaacetates 21 and 31, the pentabromide 29 and the hexabromide 32. In principle these should be convertible by elimination reactions to the desired target molecule.

Graphical Abstract
Introduction
Radialenes are cyclic cross-conjugated hydrocarbons that consist solely of semicyclic double bonds. Beginning with [3]radialene (1), prepared first by Dorko in 1965 [1], this class of basic oligoenes has been the subject of constantly growing attention by preparative and structural chemists, and – more recently – by material scientists [2-5]. Although derivatives bearing alkyl and aryl and also functionalized substituents, beginning with 1 and ending with [6]radialene (4, Scheme 1) are known for the whole series [2-5], among the parent hydrocarbons one representative is still missing: [5]radialene (3).
![[1860-5397-9-195-i1]](/bjoc/content/inline/1860-5397-9-195-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The first members of the [n]radialene series and retrosynthesis for [5]radialene (3).
Scheme 1: The first members of the [n]radialene series and retrosynthesis for [5]radialene (3).
One reason for this absence is certainly the expected low stability of 3: all its lower and higher analogs are highly reactive hydrocarbons that under normal laboratory conditions polymerize easily and/or readily undergo addition reactions with other compounds – oxygen [2,3], dienophiles [2,3], carbenes [6], and other trapping agents.
Another reason is probably the practical consideration that there are more and higher-yielding preparative routes to three-, four- and six-membered rings than to five-membered ones.
Looking at possible precursors for 3, it seems reasonable to start with a substrate that contains at least a complete five-membered carbocycle or – even better – a starting material that already displays all ten carbon atoms required in the product. For the first protocol, derivatives such as 6 are possible candidates, not least for the simple reason that in all other reported syntheses of the radialene hydrocarbons [2,3] final steps are involved that employ β-elimination processes – be it of halides, aminoxide derivatives (Cope elimination), acetates (ester pyrolysis), etc. [2,3].
When starting with derivatives of type 6 it should be noted that these compounds can exist in the form of different diastereomers; for example – as shown in 5 – all five substituents could point in the same direction (cis,cis,cis,cis-configuration). Alternatively, one (or more) of these functionalized groups could be directed in opposite directions (e.g. cis,cis,cis,trans-configuration, as in 7). Whether this stereochemical difference will have an influence on the ultimate outcome of the multiple elimination process is an open question; but it is likely to influence the rate of this multi-step transformation, which will involve increasingly reactive (unstable) intermediates.
As far as substrates are concerned that possess the complete set of necessary carbon atoms, the commercially available pentamethylcyclo-1,3-pentadiene (8) offers itself as the best starting material.
In the present publication we describe the synthesis of the two diastereomeric derivatives 5 and 7 bearing various functional groups (see below), and describe the reaction of 8 with various bromination reagents. Whenever possible we confirm the structures of intermediate compounds or byproducts by X-ray structural analysis.
Results and Discussion
cis,cis,cis,cis-1,2,3,4,5-Pentakis(hydroxymethyl)cyclopentane (16) and derivatives
The pentaalcohol 16 is a known compound, having first been described by Tolbert and co-workers in 1985 in a short communication [7]. Although in a later contribution these authors reported on the all-cis geometry (X-ray analysis) of a derivative of this compound (the pentamesylate, see below) [8]. The experimental and spectroscopic details of the original synthesis were never fully published [9]. This missing information will be presented in this publication (see Supporting Information File 1). The Tolbert synthesis of 16 is summarized in Scheme 2.
![[1860-5397-9-195-i2]](/bjoc/content/inline/1860-5397-9-195-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of cis,cis,cis,cis-1,2,3,4,5-pentakis(hydroxymethyl)cyclopentane (16) according to Tolbert [7].
Scheme 2: Preparation of cis,cis,cis,cis-1,2,3,4,5-pentakis(hydroxymethyl)cyclopentane (16) according to Tolb...
The crucial intermediate 5-[(benzyloxy)methyl]cyclopentadiene (12) was first prepared from benzyl alcohol (9) via the chloro ether 10 and cyclopentadienylthallium (11) according to a route developed by Corey and co-workers in their prostaglandin work [10]. Cycloaddition of maleic anhydride (MA) to 12 subsequently afforded the endo-adduct 13, which after esterification provided the diester 14 in excellent yield [7]. This bicyclic adduct was converted by ozonolysis followed by LAH reduction into the all-cis-derivative 15, whose benzyl ether protection group was finally removed by catalytic hydrogenation over palladium on carbon [7,8]. The full experimental details and the complete set of the spectroscopic data of 16 are collected in Supporting Information File 1.
The first successful attempt to convert the pentaalcohol into a derivative bearing better leaving groups is Tolbert´s preparation of the pentamesylate 18 (Scheme 3) [8].
![[1860-5397-9-195-i3]](/bjoc/content/inline/1860-5397-9-195-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The preparation of derivatives of 16 better suited for nucleophilic substitution and elimination.
Scheme 3: The preparation of derivatives of 16 better suited for nucleophilic substitution and elimination.
All our attempts to prepare the all-cis-pentabromide 17 from 16 failed so far, as is also shown in Scheme 3. Only in one case was a halogenated derivative obtained in poor yield: treatment of 16 with triphenylphosphine–bromine in carbon tetrachloride provided the tribromo ether derivative 19 as a minor component. Surprisingly, this had the cis,cis,cis,trans-configuration of the substituents at the five-membered ring, as was shown by single crystal X-ray analysis (Figure 1).
![[1860-5397-9-195-1]](/bjoc/content/figures/1860-5397-9-195-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of 19 in the crystal; ellipsoids represent 50% probability levels.
Figure 1: Structure of 19 in the crystal; ellipsoids represent 50% probability levels.
It is unclear at the present time whether the correspondingly configurated pentaalcohol was already present in the starting material 16 as a side-product, which had been overlooked in previous studies, or whether epimerization took place during the attempted conversion of the pentaalcohol to the pentabromide 17. The complete, structure-confirming spectroscopic and analytical data of 19 can be found in Supporting Information File 1.
In closing this section on the preparation of bromine derivatives of 16, it should also be mentioned that attempts to prepare the corresponding pentachloride (by treatment with either SOCl2 or PCl3) and pentaiodide (red P/iodine) also failed. In all these experiments complex product mixtures resulted, the GC–MS analysis of which demonstrated that only partially halogenated derivatives of 16 had been produced; their chromatographic separation failed.
In order to avoid working in solution and subjecting the presumably reactive 5 to extended work-up and purification conditions, we next tried to prepare the xanthate 20 and the acetate 21 from 16, derivatives that could be subjected to flash vacuum pyrolysis (Scheme 4).
![[1860-5397-9-195-i4]](/bjoc/content/inline/1860-5397-9-195-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of the pentaacetate 21 from 16.
Scheme 4: Preparation of the pentaacetate 21 from 16.
Whereas all our attempts to synthesize 20 failed (see Supporting Information File 1), the pentaester 21 was obtained under standard conditions from 16 by treatment with excess acetyl chloride in excellent yield; its spectroscopic data can be found in the Supporting Information File 1.
cis,cis,cis,trans-1,2,3,4,5- (26), cis,cis,trans,cis-1,2,3,4,5-Pentakis(methoxycarbonyl)cyclopentane (27) and several of their derivatives
For the other two routes to precursors of [5]radialene (3) discussed here, the ten carbon atoms of the desired final product were available already at a very early stage of the synthesis.
The first route dates back to studies by Diels, Alder and co-workers [11,12], carried out in the early 1930s and 40s, which were taken up later by Le Goff and his students [13]. These pioneers demonstrated that, on treatment of dimethyl acetylenedicarboxylate (22) with dimethyl malonate (23) under the catalytic influence of pyridinium acetate, the fully substituted cycloheptadienes 24 and 25 (Scheme 5) were produced; both authors also proposed reaction mechanisms for this complex transformation that can be considered as reasonable even today; for the details the reader is referred to the original literature [12,13].
![[1860-5397-9-195-i5]](/bjoc/content/inline/1860-5397-9-195-i5.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of the cycloheptadiene octaesters 24/25 according to Diels [11] and Le Goff [13], respectively, followed by ring contraction to the cyclopentadiene pentaester 28.
Scheme 5: Preparation of the cycloheptadiene octaesters 24/25 according to Diels [11] and Le Goff [13], respectively,...
The reproduction of this protocol posed no problems (see Supporting Information File 1). The only difference we noted between our results and those reported in the chemical literature concerns the ratio of 24 to 25. Whereas Diels reported a ratio of 3:1, and in Le Goff´s hands it was as much as 15:1, we observed the formation of 24 as a minor product (24:25 = 1:8). One reason for this difference could be a pericyclic 1,5-hydrogen shift between the two valence isomers generating different ratios under different preparation and work-up conditions. Another equilibration mechanism could involve a base-induced 1,5-hydrogen shift. Since the mechanism of formation of 24 and 25 was not the main concern of our investigations, we did not clarify these points. The differentiation between the symmetrical 24 and the non-symmetrical 25 by 1H NMR spectroscopy is easy and was achieved by Le Goff. More complete and up-to-date spectroscopic data of the two compounds are summarized in Supporting Information File 1. Still, the early workers [11-13] did not distinguish between a possible meso-structure (cis-orientation of allylic hydrogen atoms) and the chiral (C2-symmetrical) configuration (trans-orientation of hydrogen atoms) of 24. Fortunately, recrystallization of a 24/25-mixture from diethyl ether provided single crystals of 24 suitable for an X-ray structure investigation which proved the chiral configuration (Figure 2).
![[1860-5397-9-195-2]](/bjoc/content/figures/1860-5397-9-195-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of 24 in the crystal; ellipsoids represent 30% probability levels.
Figure 2: Structure of 24 in the crystal; ellipsoids represent 30% probability levels.
The ring contraction of a mixture of 24/25, following the procedure given by Le Goff [13], proceeded as expected and provided the potassium salt of 28 in acceptable yields (60%). From this, free 1,2,3,4,5-pentakis(methoxycarbonyl)cyclopenta-1,3-diene 28 was liberated by hydrochloric acid treatment in excellent yield (>90%, Scheme 5).
Finally, the hydrogenation of 28 over Pd/C in methanol furnished a mixture of the saturated pentaesters 26 and 27 (ratio 4:1, capillary GC–MS-analysis) in practically quantitative yield (98%). Although we were unable to separate the two components quantitatively by column chromatography on silica gel, the main isomer began to crystallize partially from the highly viscous hydrogenation mixture after standing for several months. The crystals obtained were suitable for X-ray structural analysis, which showed that this isomer has the cis,cis,cis,trans-geometry, i.e. 26 (Figure 3).
![[1860-5397-9-195-3]](/bjoc/content/figures/1860-5397-9-195-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Structure of 26 in the crystal; ellipsoids represent 30% probability levels.
Figure 3: Structure of 26 in the crystal; ellipsoids represent 30% probability levels.
The structural assignment of the minor hydrogenation product as 27, which could only be obtained in enriched form (ratio 26:27 ca. 1:1) is based on the following considerations. Altogether there exist three hydrogenation products of 28: the two shown (26, 27) and also the all-cis-configurated pentaester, presumably the most highly strained of the three isomers. For symmetry reasons this compound should display two sharp signals in its 1H NMR spectrum: one for the tertiary hydrogen atom at the ring and one for the methyl group of its five identical ester groups (ratio 1:3). It should not be difficult to identify these signals in the proton spectrum of a mixture of hydrogenation products because of their relative intensities. Although a large number of signals can indeed be recognized in the corresponding chemical shift regions, none displays an extraordinary intensity. It is hence unlikely that the all-cis-pentaester is produced in significant amounts on hydrogenation.
As far as derivatives of 26 and 27 are concerned, we always worked with the mixture of the two diastereomers (Scheme 6).
![[1860-5397-9-195-i6]](/bjoc/content/inline/1860-5397-9-195-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Derivatives derived from the pentaester mixture 26/27.
Scheme 6: Derivatives derived from the pentaester mixture 26/27.
Reduction of 26/27 with lithium aluminum hydride in either diethyl ether or THF provided the diastereomeric pentaalcohols 30. The yield of the process is probably higher than the finally isolated 50%. Both isomers are readily soluble in water, and to obtain the pure mixture the crude reduction product was first extracted with diethyl ether or THF. This leaves the inorganic salts and the pentaalcohols as a residual solid. When this is extracted with refluxing methanol or (preferably) isopropanol the desired reduction product 30 slowly dissolves, leaving the salts behind. The removal of the solvent from 30 is difficult and requires extended pumping under high vacuum (2 weeks). Even then the residue shows traces of the originally added solvent (by NMR analysis).
The exact structure assignment of 30 is hampered by the fact that no pure isomer was available. We assume, however, that the isomeric composition is not changed by the LAH reduction (i.e. 4:1 as in the case of the esters 26/27). The signals for the cis,cis,cis,trans-pentaalcohol are clearly discernible. The 1H NMR spectrum displays three signal groups from δ = 1.63–2.52 in 2:2:1 ratio for the ring protons. In the 13C NMR spectrum the ring methine carbon atoms absorb at 45.5, 46.0, and 46.7 ppm, whereas the primary carbon atoms are registered at δ = 60.0, 63.2, and 65.5 ppm. The signals of the minor isomer(s) can hardly be differentiated from the noise.
The bromination of 30 proceeded with varying results; most of the methods that were unsuccessfully used above in the case of 16 also fail for its isomer (HBr, PBr3, SOBr2 in different halogenated solvents). However, with the triphenylphosphine–bromine complex, a mixture of the pentabromides 29 was obtained in very good yield (73%). The superior results in comparison with 16 probably can be rationalized by a decreased steric shielding of the hydroxymethyl groups in 30, but are probably also associated with the different solubility of the pentaalcohol mixture. Likewise, the pentaacetate 31 can be obtained from 30 in excellent yield (Scheme 5). As in the case of 16, no xanthate (see above) could be prepared from 30.
Bromination of 1,2,3,4,5-pentamethylcyclopenta-1,3-diene (8)
The other substrate that has all the carbon atoms of the final [5]radialene (3) already in place is 1,2,3,4,5-pentamethylcyclopenta-1,3-diene (8). Formally this hydrocarbon merely has to be dehydrogenated three times to provide the target molecule. To accomplish this goal we first had to replace these hydrogen atoms by a better leaving group, e.g. bromine as in Scheme 7.
![[1860-5397-9-195-i7]](/bjoc/content/inline/1860-5397-9-195-i7.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Bromination of 1,2,3,4,5-pentamethylcyclopenta-1,3-diene (8).
Scheme 7: Bromination of 1,2,3,4,5-pentamethylcyclopenta-1,3-diene (8).
After trying numerous different protocols (Br2 addition, NBS in different concentrations and in different solvents, 1,3-dibromo-5,5-dimethylhydantoin) [14] we finally found that excess NBS (6 equivalents) and refluxing in tetrachloromethane at 80 °C gave the best results. Under these conditions the hexabromide 32 was produced in 45% yield. The structure of this cyclopentadiene derivative was derived from its spectroscopic data (see Supporting Information File 1) and also by an X-ray structural analysis (Figure 4).
![[1860-5397-9-195-4]](/bjoc/content/figures/1860-5397-9-195-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Structure of 32 in the crystal; ellipsoids represent 50% probability levels.
Figure 4: Structure of 32 in the crystal; ellipsoids represent 50% probability levels.
A rationalization of the route from 8 to 32 is also summarized in Scheme 7. Since in all our bromination experiments we isolated small amounts of a hydrocarbon that, according to mass spectral analysis, has the formula C20H30, i.e. is a dimer of the starting material, we assume that the permethylcyclopentadienyl radical 33 plays an important role in the bromination reaction. This could not only lead to the bromo derivative 34 in which the final ring hydrogen atom has been substituted, but also to a mixture of hexabromides among which the hexabromide 32 is the major reaction product.
With several derivatives (18 [7,8], 21, 29, 31, and 32) that could lead to [5]radialene (3) in hand, a synthesis of this long missing hydrocarbon is now within sight.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 309.8 KB | Download |
References
-
Dorko, E. A. J. Am. Chem. Soc. 1965, 87, 5518–5520. doi:10.1021/ja00951a067
Return to citation in text: [1] -
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573
Return to citation in text: [1] [2] -
Hopf, H. Angew. Chem. 2012, 124, 12111–12113. doi:10.1002/ange.201206101
Angew. Chem., Int. Ed. 2012, 51, 11945–11947. doi:10.1002/anie.201206101. The number of known compounds possessing a radialene-type distribution of their π-electrons increases quickly once heteroatoms (O, S,-NR etc.) are introduced into the carbon skeleton; for a short review on recent developments in heteroradialene chemistry.
Return to citation in text: [1] [2] -
Trabert, L.; Hopf, H.; Schomburg, D. Chem. Ber. 1981, 114, 2405–2414. doi:10.1002/cber.19811140707
Return to citation in text: [1] -
Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030
Return to citation in text: [1] [2] [3] [4] [5] -
Brock, C. P.; Gregory, J. C.; Tolbert, L. M. Acta Crystallogr., Sect. C 1986, 42, 1063–1066. doi:10.1107/S0108270186093460
Return to citation in text: [1] [2] [3] [4] -
L. M. Tolbert (Georgia Institute of Technology), private communication to H. Hopf.
Return to citation in text: [1] -
Corey, E. J.; Koelliker, U.; Neuffer, J. J. Am. Chem. Soc. 1971, 93, 1489–1490. doi:10.1021/ja00735a031
Return to citation in text: [1] -
Diels, O.; Alder, K. Justus Liebigs Ann. Chem. 1932, 498, 16–49. doi:10.1002/jlac.19324980103
Return to citation in text: [1] [2] [3] -
Diels, O. Ber. Dtsch. Chem. Ges. 1942, 75, 1452–1467. doi:10.1002/cber.19420751206
Return to citation in text: [1] [2] [3] -
Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043
Return to citation in text: [1] [2] [3] [4] [5] -
Oakes, V.; Rydon, H. N.; Undheim, K. J. Chem. Soc. 1962, 4678–4685. doi:10.1039/jr9620004678
Return to citation in text: [1]
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 7. | Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030 |
| 8. | Brock, C. P.; Gregory, J. C.; Tolbert, L. M. Acta Crystallogr., Sect. C 1986, 42, 1063–1066. doi:10.1107/S0108270186093460 |
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 8. | Brock, C. P.; Gregory, J. C.; Tolbert, L. M. Acta Crystallogr., Sect. C 1986, 42, 1063–1066. doi:10.1107/S0108270186093460 |
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 4. | Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573 |
| 5. |
Hopf, H. Angew. Chem. 2012, 124, 12111–12113. doi:10.1002/ange.201206101
Angew. Chem., Int. Ed. 2012, 51, 11945–11947. doi:10.1002/anie.201206101. The number of known compounds possessing a radialene-type distribution of their π-electrons increases quickly once heteroatoms (O, S,-NR etc.) are introduced into the carbon skeleton; for a short review on recent developments in heteroradialene chemistry. |
| 10. | Corey, E. J.; Koelliker, U.; Neuffer, J. J. Am. Chem. Soc. 1971, 93, 1489–1490. doi:10.1021/ja00735a031 |
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 4. | Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573 |
| 5. |
Hopf, H. Angew. Chem. 2012, 124, 12111–12113. doi:10.1002/ange.201206101
Angew. Chem., Int. Ed. 2012, 51, 11945–11947. doi:10.1002/anie.201206101. The number of known compounds possessing a radialene-type distribution of their π-electrons increases quickly once heteroatoms (O, S,-NR etc.) are introduced into the carbon skeleton; for a short review on recent developments in heteroradialene chemistry. |
| 7. | Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030 |
| 7. | Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030 |
| 9. | L. M. Tolbert (Georgia Institute of Technology), private communication to H. Hopf. |
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 7. | Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030 |
| 2. |
Hopf, H.; Maas, G. Angew. Chem. 1992, 104, 953–977. doi:10.1002/ange.19921040804
Angew. Chem. Int., Ed. Engl. 1992, 31, 931–954. doi:10.1002/anie.199209313. The first comprehensive review on [n]radialenes and their derivatives. |
| 3. | Maas, G.; Hopf, H. In The Chemistry of Functional Groups; Patai, S.; Rappoport, Z., Eds.; The Chemistry of Dienes and Polyenes, Vol. 1; J. Wiley & Sons: Chichester, 1997; pp 927 ff. |
| 6. | Trabert, L.; Hopf, H.; Schomburg, D. Chem. Ber. 1981, 114, 2405–2414. doi:10.1002/cber.19811140707 |
| 8. | Brock, C. P.; Gregory, J. C.; Tolbert, L. M. Acta Crystallogr., Sect. C 1986, 42, 1063–1066. doi:10.1107/S0108270186093460 |
| 12. | Diels, O. Ber. Dtsch. Chem. Ges. 1942, 75, 1452–1467. doi:10.1002/cber.19420751206 |
| 13. | Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043 |
| 11. | Diels, O.; Alder, K. Justus Liebigs Ann. Chem. 1932, 498, 16–49. doi:10.1002/jlac.19324980103 |
| 12. | Diels, O. Ber. Dtsch. Chem. Ges. 1942, 75, 1452–1467. doi:10.1002/cber.19420751206 |
| 13. | Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043 |
| 14. | Oakes, V.; Rydon, H. N.; Undheim, K. J. Chem. Soc. 1962, 4678–4685. doi:10.1039/jr9620004678 |
| 7. | Tolbert, L. M.; Gregory, J. C.; Brock, C. P. J. Org. Chem. 1985, 50, 548. doi:10.1021/jo00204a030 |
| 8. | Brock, C. P.; Gregory, J. C.; Tolbert, L. M. Acta Crystallogr., Sect. C 1986, 42, 1063–1066. doi:10.1107/S0108270186093460 |
| 11. | Diels, O.; Alder, K. Justus Liebigs Ann. Chem. 1932, 498, 16–49. doi:10.1002/jlac.19324980103 |
| 12. | Diels, O. Ber. Dtsch. Chem. Ges. 1942, 75, 1452–1467. doi:10.1002/cber.19420751206 |
| 13. | Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043 |
| 13. | Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043 |
| 11. | Diels, O.; Alder, K. Justus Liebigs Ann. Chem. 1932, 498, 16–49. doi:10.1002/jlac.19324980103 |
| 13. | Le Goff, E.; LaCount, R. B. J. Org. Chem. 1964, 29, 423–427. doi:10.1021/jo01025a043 |
© 2013 Kelch et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)