Abstract
Oxabicyclic alkenes can react with electron-deficient terminal alkynes in the presence of a gold catalyst under mild conditions, affording the corresponding addition products in moderate yields. When using alkynyl esters as substrates, the (Z)-acrylate derivatives are obtained. Using but-3-yn-2-one (ethynyl ketone) as a substrate, the corresponding addition product is obtained with (E)-configuration. The proposed mechanism of these reactions is also discussed.

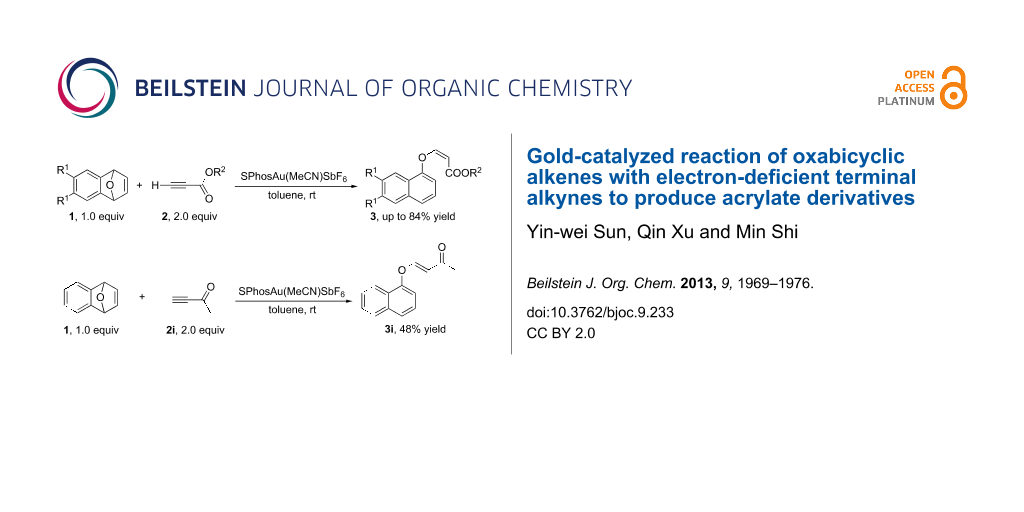
Graphical Abstract
Introduction
Oxabicyclic alkenes are common intermediates in organic synthesis since these compounds can be easily prepared and have a high reactivity for further transformations [1-8]. For example, they are often used to construct substituted tetrahydronaphthalene skeletons in the presence of metal catalysts such as Pd [9,10], Ir [11-15], Rh [16-21] and Cu [22]. However, their reactivity in the presence of gold catalysts has been rarely reported [23]. It is well known that gold catalysts have different catalytic abilities compared with other transition metals [24]. Moreover, gold-catalyzed chemical transformations have made significant progress during the last 5 years [25-56]. Many gold complexes have been proved to be efficient catalysts in C–C [33-48] bond or C–X (X = heteroatom) [49-56] bond forming reactions. Our group has a long-standing interest in gold-catalyzed C–C [57-61] or C–X bond [62-67] formation reactions. So far, we have reported a variety of gold-catalyzed intramolecular rearrangements with highly strained small rings for C–C or C–X bond formations [57-59,62-68]. Based on these previous findings, we envisaged that oxabicyclic alkenes could also react with electron-deficient alkynes in the presence of gold catalysts to generate a new C–C or C–O bond thereby releasing the oxabicyclic alkenes of their ring strain. In this paper, we report the formation of (Z)-acrylate derivatives in the gold catalyzed intermolecular reaction of oxabicyclic alkenes with electron-deficient terminal alkynes under mild conditions [69-76] (Scheme 1).
![[1860-5397-9-233-i1]](/bjoc/content/inline/1860-5397-9-233-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Gold-catalyzed reactions of oxabicyclic alkenes with electron-deficient terminal alkynes.
Scheme 1: Gold-catalyzed reactions of oxabicyclic alkenes with electron-deficient terminal alkynes.
Results and Discussion
To generate a new C–O bond in the reaction of oxabicyclic alkene 1a with electron-deficient terminal alkyne 2a, we first used PPh3AuCl as a catalyst, AgSbF6 as an additive, and toluene as a solvent to examine the reaction outcome. Acrylate derivative 3a was formed with (Z)-configuration in 11% yield (Table 1, entry 1). In this reaction, naphthalen-1-ol was also obtained with 44% yield as the major product. The usage of IPrAuCl, dppb(AuCl)2, (p-FC6H4)3PAuCl, DPE-phos(AuCl)2, Me3PAuCl and Cy3PAuCl as gold catalysts did not significantly improve the yield of 3a (Table 1, entries 2–7). In these cases, the maximum yield of 3a was 34% when the gold complex Cy3PAuCl coordinated by an electron-rich phosphine ligand was used as a catalyst (Table 1, entry 7). In order to further improve the yield of 3a, we employed gold complex 6 (Figure 1) coordinated by a sterically bulky and electron-rich biaryl phosphine-type ligand as a catalyst, affording 3a in 40% yield (Table 1, entry 8). In the absence of AgSbF6, no reaction occurred (Table 1, entry 9). The usage of AgSbF6 as a catalyst produced naphthalen-1-ol (5) as the major product (Table 1, entry 10). Next, we further screened the reaction conditions with gold complex 6 as a catalyst. When using AgOTs or CF3CO2Ag as a silver additive, we did not obtain any of the desired products (Table 1, entries 12 and 13), whereas the usage of AgNTf2 as a silver additive afforded 3a in 32% yield (Table 1, entry 11). AgOTf was not an effective silver additive, giving 3a in 20% yield (Table 1, entry 14). Utilization of the already prepared electrophilic cationic phosphinogold(I) complexes XPhosAuNTf2 and XPhosAu(MeCN)SbF6 as gold catalysts slightly increased the yield of 3a to 33% and 45% yields, respectively (Table 1, entries 15 and 16). The examination of solvent effects revealed that toluene was the best solvent (Table 1, entries 17–22). Adding 4 Å MS into the reaction system, 3a was obtained in only 10% yield (Table 1, entry 23).
Table 1: Initial screening of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-233-i5.png?max-width=637&scale=1.0)
|
||||
| Entrya | Au cat. | Ag salt | Solvent | Yieldb (%) 3a |
|---|---|---|---|---|
| 1c | Ph3PAuCl | AgSbF6 | toluene | 11 |
| 2 | IPrAuCl | AgSbF6 | toluene | 19 |
| 3 | dppb(AuCl)2 | AgSbF6 | toluene | trace |
| 4 | (p-FC6H4)3PAuCl | AgSbF6 | toluene | 26 |
| 5 | DPE-phos(AuCl)2 | AgSbF6 | toluene | N.R. |
| 6 | Me3PAuCl | AgSbF6 | toluene | N.R. |
| 7 | Cy3PAuCl | AgSbF6 | toluene | 34 |
| 8 | 6 | AgSbF6 | toluene | 40 |
| 9 | 6 | – | toluene | N.R. |
| 10c | None | AgSbF6 | toluene | trace |
| 11 | 6 | AgNTf2 | toluene | 32 |
| 12 | 6 | AgOTs | toluene | N.R. |
| 13 | 6 | CF3COOAg | toluene | N.R. |
| 14 | 6 | AgOTf | toluene | 20 |
| 15 | XPhosAuNTf2 | – | toluene | 33 |
| 16 | XPhosAu(MeCN)SbF6 | – | toluene | 45 |
| 17 | XPhosAu(MeCN)SbF6 | – | CH3CN | trace |
| 18 | XPhosAu(MeCN)SbF6 | – | CH3NO2 | N.R. |
| 19 | XPhosAu(MeCN)SbF6 | – | Et2O | N.R. |
| 20 | XPhosAu(MeCN)SbF6 | – | THF | complex |
| 21 | XPhosAu(MeCN)SbF6 | – | DCM | 25 |
| 22 | XPhosAu(MeCN)SbF6 | – | DCE | 28 |
| 23d | XPhosAu(MeCN)SbF6 | – | toluene | 10 |
aThe reaction was carried out on a 0.2 mmol scale in solvent (1.0 mL). The ratio of 1a/2a was 1:2. bYield determined by 1H NMR by using 1-iodo-2-methoxybenzene (4) as an internal standard. cNaphthalen-1-ol (5) was the major product. d50 mg of 4 Å MS was added to the reaction system.
![[1860-5397-9-233-1]](/bjoc/content/figures/1860-5397-9-233-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Gold complexes used in this reaction.
Figure 1: Gold complexes used in this reaction.
Since the yield of 3a was still low, we next tried to improve the yield of 3a by deploying different ligands, Ag salts, solvents and temperature. The results are summarized in Table 2. At first, we examined many other gold(I) phosphane complexes with dialkylbiarylphosphane ligands (Figure 1) by using AgSbF6 as an additive and toluene as a solvent. No reaction occurred when gold(I) phosphane complexes 8–10 (Figure 1) were used as catalysts under identical conditions (Table 2, entries 2–4). Furthermore, the usage of gold(I) phosphane complexes 7, 11, 13 and 14 (Figure 1) as catalysts gave 3a in 10–29% yields (Table 2, entries 1, 5, 7 and 8). Gold complex 12 (Figure 1) with an electron-rich biphenylphosphine ligand was identified as the best catalyst, giving 3a in 67% yield (Table 2, entry 6). We attempted to further optimize the reaction conditions by using SPhosAuCl 12 as a catalyst and AgNTf2 or AgSbF6 as an additive and obtained 3a in 66% and 67% yields, respectively (Table 2, entries 6 and 9). However, the use of AgOTf or AgBF4 as an additive afforded 3a in 37% and 11% yields, respectively (Table 2, entries 10 and 11). Employment of the prepared electrophilic cationic phosphinogold(I) complex SPhosAu(MeCN)SbF6 as a catalyst gave 3a in 78% NMR based yield and 67% isolated yield (Table 2, entry 12). The phosphinogold(I) complex SPhosAuNTf2 produced 3a in 53% yield under the standard conditions (Table 2, entry 13). The examination of solvent effects disclosed that toluene was the best solvent (Table 2, entries 14–16). Either increasing or decreasing the reaction temperature did not further improve the reaction outcome (Table 2, entries 17–20). Careful screening of the reaction conditions led to the conclusion that the reaction should be carried out in toluene at room temperature with SPhosAu(MeCN)SbF6 as the catalyst (Table 2, entry 12).
Table 2: Further screening of the reaction conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-233-i6.png?max-width=637&scale=1.0)
|
|||||
| entrya | Au cat. | Ag salt | solvent | T (°C) | Yieldb (%) 3a |
|---|---|---|---|---|---|
| 1c | 7 | AgSbF6 | toluene | rt | 11 |
| 2 | 8 | AgSbF6 | toluene | rt | N.R. |
| 3 | 9 | AgSbF6 | toluene | rt | trace |
| 4 | 10 | AgSbF6 | toluene | rt | N.R. |
| 5 | 11 | AgSbF6 | toluene | rt | 10 |
| 6 | 12 | AgSbF6 | toluene | rt | 67 |
| 7 | 13 | AgSbF6 | toluene | rt | 20 |
| 8 | 14 | AgSbF6 | toluene | rt | 29 |
| 9 | 12 | AgNTf2 | toluene | rt | 66 (59)c |
| 10 | 12 | AgOTf | toluene | rt | 37 |
| 11 | 12 | AgBF4 | toluene | rt | 11 |
| 12 | SPhosAu(MeCN)SbF6 | – | toluene | rt | 78 (67)c |
| 13 | SPhosAuNTf2 | – | toluene | rt | 53 |
| 14 | SPhosAu(MeCN)SbF6 | – | DCM | rt | 49 |
| 15 | SPhosAu(MeCN)SbF6 | – | DCE | rt | 55 |
| 16 | SPhosAu(MeCN)SbF6 | – | CHCl3 | rt | 45 |
| 17d | SPhosAu(MeCN)SbF6 | – | toluene | 0 | 50 |
| 18 | SPhosAu(MeCN)SbF6 | – | toluene | 40 | 45 |
| 19 | SPhosAu(MeCN)SbF6 | – | toluene | 10 | 59 |
| 20 | SPhosAu(MeCN)SbF6 | – | toluene | 30 | 70 |
aThe reaction was carried out on a 0.2 mmol scale in solvent (1.0 mL) and the ratio of 1a/2a was 1/2. bYield determined by 1H NMR by using 1-iodo-2-methoxybenzene 4 as an internal standard. cIsolated yield in parentheses.
Having identified the optimal conditions, we next examined the substrate scope of this reaction. We found that only the usage of ethynylbenzene and dimethyl but-2-ynedioate as substrates did not afford any of the desired products (Table 3, entries 6 and 9). In all other cases, the reactions proceeded smoothly to give the desired products in moderate to good yields (Table 2, entries 1–5, 7 and 8). The introduction of electron-donating substituents on the benzene ring impaired the reaction outcome (Table 3, entries 1 and 7). Increasing the steric hindrance of the ester group improved the yields of 3 (Table 3, entries 4 and 5). The usage of but-3-yne-2-one (terminal alkyne ketone) 2i as a substrate gave the corresponding 3i with (E)-configuration in 48% yield (Scheme 2).
Table 3: Substrate scope of the reactions with SPhosAu(MeCN)SbF6 as a catalyst.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-233-i7.png?max-width=637&scale=1.0)
|
|||
| entrya | R1 | R2, R3 | Yieldb (%) 3 |
|---|---|---|---|
| 1 | Me | COOMe, H | 3b, 42 |
| 2 | F | COOMe, H | 3c, 66 |
| 3 | Br | COOMe, H | 3d, 76 |
| 4 | H | COOEt, H | 3e, 54 |
| 5 | H | COOt-Bu, H | 3f, 84 |
| 6 | H | Ph, H | trace |
| 7 | Me | COOt-Bu, H | 3g, 58 |
| 8 | Br | COOt-Bu, H | 3h, 72 |
| 9 | H | COOMe, COOMe | N.R. |
aThe reaction was carried out on a 0.2 mmol scale in solvent (1.0 mL). The ratio of 1/2 was 1/2. bIsolated yield.
![[1860-5397-9-233-i2]](/bjoc/content/inline/1860-5397-9-233-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The reaction with terminal alkyne 2i as a substrate.
Scheme 2: The reaction with terminal alkyne 2i as a substrate.
Since naphthalene-1-ol (5) was obtained in this reaction, we used naphthalene-1-ol (5) as a substrate and carried out the reaction under the optimal conditions to clarify whether the reaction proceeded through naphthalene-1-ol. The formation of 3a could not be observed, suggesting that naphthalene-1-ol is not the intermediate in this reaction (Scheme 3).
![[1860-5397-9-233-i3]](/bjoc/content/inline/1860-5397-9-233-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The reaction with naphthalen-1-ol (5) as a substrate.
Scheme 3: The reaction with naphthalen-1-ol (5) as a substrate.
Based on the previously established mechanistic model [23,77], we propose the following pathway for the formation of acrylate derivatives 3a and 3i (Scheme 4). In the presence of cationic phosphinogold(I) complex, a cationic intermediate A is formed by a regioselective opening of the oxygen bridge in substrate 1a. Intermediate A releases a proton to afford intermediate B. Intermediate B attacks methyl propiolate, which is activated by the gold catalyst, to generate gold vinyl complex C. In intermediate C, the ester group and the naphthalene ring are on the same side, yielding the final product 3a with (Z)-configuration via protodeauration. The alkyne ketone 3i is more electron-deficient and more reactive than methyl propiolate and it is more difficult to coordinate by the gold complex. Therefore, with alkyne ketone 2i as a substrate, intermediate B attacks non-coordinated alkyne ketone 2i in a cis-addition manner to generate gold vinyl complex D. In intermediate D, the carbonyl group and the naphthalene ring are on opposite sides, affording product 3i with (E)-configuration by protodeauration.
![[1860-5397-9-233-i4]](/bjoc/content/inline/1860-5397-9-233-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The proposed mechanism for Au(I)-catalyzed reaction.
Scheme 4: The proposed mechanism for Au(I)-catalyzed reaction.
Conclusion
In summary, we have developed a novel method to synthesize acrylate derivatives from oxabicyclic alkenes and electron-deficient terminal alkynes in toluene in moderate to good yields in the presence of the gold catalyst SPhosAu(MeCN)SbF6 under mild conditions. Efforts are in progress to elucidate the mechanistic details of this reaction and to disclose its scope and limitations.
Experimental
General remarks
Dichloromethane was freshly distilled from calcium hydride; THF and toluene were distilled from sodium under an argon atmosphere. 1H NMR, 13C NMR and 19F NMR spectra were recorded on a Bruker AM-400 spectrometer. Infrared spectra were recorded on a Perkin-Elmer PE-983 spectrometer with absorption in cm−1. Flash column chromatography was performed by using 300–400 mesh silica gel. For thin-layer chromatography (TLC), silica gel plates (Huanghai GF254) were used. Mass spectra were recorded by ESI, and HRMS were measured on a HP-5989 instrument.
General procedure for the reaction catalyzed by Au(I) catalysts
Into an oven-dried reaction flask under Ar gas protection was added oxabicyclic alkene (0.2 mmol), Au catalyst (0.001 mmol), methyl propiolate (0.4 mmol) and toluene (1.0 mL). The reaction mixture was stirred at room temperature normally overnight. After complete consumption of the starting materials, monitored by TLC, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of compounds. | ||
| Format: PDF | Size: 499.2 KB | Download |
Acknowledgements
Financial support from the Shanghai Municipal Committee of Science and Technology (08dj1400100-2), the Fundamental Research Funds for the Central Universities, the National Basic Research Program of China (973)-2010CB833302, the Fundamental Research Funds for the Central Universities, and the National Natural Science Foundation of China (21072206, 20902019, 20472096, 20872162, 20672127, 20732008, 20821002, and 20702013) are gratefully acknowledged.
References
-
Tsui, G. C.; Tsoung, J.; Dougan, P.; Lautens, M. Org. Lett. 2012, 14, 5542–5545. doi:10.1021/ol302646a
Return to citation in text: [1] -
Mannathan, S.; Cheng, C.-H. Chem. Commun. 2013, 49, 1557–1559. doi:10.1039/C2CC38001C
Return to citation in text: [1] -
Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106–6108. doi:10.1039/C2CC31880F
Return to citation in text: [1] -
Jack, K.; Fatila, E.; Hillis, C.; Tam, W. Synth. Commun. 2013, 43, 1181–1187. doi:10.1080/00397911.2011.626140
Return to citation in text: [1] -
Endo, K.; Tanaka, K.; Ogawa, M.; Shibata, T. Org. Lett. 2011, 13, 868–871. doi:10.1021/ol102928q
Return to citation in text: [1] -
Madan, S.; Cheng, C.-H. J. Org. Chem. 2006, 71, 8312–8315. doi:10.1021/jo061477h
Return to citation in text: [1] -
Lautens, M.; Rovis, T. J. Org. Chem. 1997, 62, 5246–5247. doi:10.1021/jo971115x
Return to citation in text: [1] -
Barluenga, J.; Rodriguez, F.; Alvarez-Rodrigo, L.; Zapico, J. M.; Fañanás, F. J. Chem.–Eur. J. 2004, 10, 109–116. doi:10.1002/chem.200305374
Return to citation in text: [1] -
Ge, G.-C.; Mo, D.-L.; Ding, C.-H.; Dai, L.-X.; Hou, X.-L. Org. Lett. 2012, 14, 5756–5759. doi:10.1021/ol302586m
Return to citation in text: [1] -
Huang, X.-J.; Mo, D.-L.; Ding, C.-H.; Hou, X.-L. Synlett 2011, 943–946. doi:10.1055/s-0030-1259716
Return to citation in text: [1] -
Hu, J.; Yang, Q.; Yu, L.; Xu, J.; Liu, S.; Huang, C.; Wang, L.; Zhou, Y.; Fan, B. Org. Biomol. Chem. 2013, 11, 2294–2301. doi:10.1039/C3OB27382B
Return to citation in text: [1] -
Hu, J.; Yang, Q.; Xu, J.; Huang, C.; Fan, B.; Wang, J.; Lin, C.; Bian, Z.; Chan, A. S. C. Org. Biomol. Chem. 2013, 11, 814–820. doi:10.1039/C2OB26775F
Return to citation in text: [1] -
Cheng, H.; Yang, D. J. Org. Chem. 2012, 77, 9756–9765. doi:10.1021/jo3018507
Return to citation in text: [1] -
Yang, D.; Long, Y.; Zhang, J.; Zeng, H.; Wang, S.; Li, C. Organometallics 2010, 29, 3477–3480. doi:10.1021/om100384q
Return to citation in text: [1] -
Fan, B.-M.; Li, X.-J.; Peng, F.-Z.; Zhang, H.-B.; Chan, A. S. C.; Shao, Z.-H. Org. Lett. 2010, 12, 304–306. doi:10.1021/ol902574c
Return to citation in text: [1] -
Tsui, G. C.; Dougan, P.; Lautens, M. Org. Lett. 2013, 15, 2652–2655. doi:10.1021/ol4009393
Return to citation in text: [1] -
Tsui, G. C.; Ninnemann, N. M.; Hosotani, A.; Lautens, M. Org. Lett. 2013, 15, 1064–1067. doi:10.1021/ol4000668
Return to citation in text: [1] -
Zhu, J.; Tsui, G. C.; Lautens, M. Angew. Chem., Int. Ed. 2012, 51, 12353–12356. doi:10.1002/anie.201207356
Return to citation in text: [1] -
Tsui, G. C.; Lautens, M. Angew. Chem., Int. Ed. 2012, 51, 5400–5404. doi:10.1002/anie.201200390
Return to citation in text: [1] -
Preetz, A.; Kohrt, C.; Drexler, H.-J.; Torrens, A.; Buschmann, H.; Lopez, M. G.; Heller, D. Adv. Synth. Catal. 2010, 352, 2073–2080. doi:10.1002/adsc.201000236
Return to citation in text: [1] -
Long, Y.; Zhao, S.; Zeng, H.; Yang, D. Catal. Lett. 2010, 138, 124–133. doi:10.1007/s10562-010-0383-3
Return to citation in text: [1] -
Bos, P. H.; Rudolph, A.; Pérez, M.; Fañanás-Mastral, M.; Harutyunyan, S. R.; Feringa, B. L. Chem. Commun. 2012, 48, 1748–1750. doi:10.1039/C2CC16855C
Return to citation in text: [1] -
Sawama, Y.; Kawamoto, K.; Satake, H.; Krause, N.; Kita, Y. Synlett 2010, 2151–2155. doi:10.1055/s-0030-1258528
Return to citation in text: [1] [2] -
Brazeau, J.-F.; Zhang, S.; Colomer, I.; Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2012, 134, 2742–2749. doi:10.1021/ja210388g
Return to citation in text: [1] -
Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/C1CS15088J
Return to citation in text: [1] -
Sethofer, S. G.; Mayer, T.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8276–8277. doi:10.1021/ja103544p
Return to citation in text: [1] -
Rudolph, M. In Modern Gold Catalyzed Synthesis; Hashmi, A. S. K.; Toste, F. D., Eds.; Wiley: Weinheim, Germany, 2012; pp 331–362.
Return to citation in text: [1] -
Hubbert, C.; Hashmi, A. S. K. In Modern Gold Catalyzed Synthesis; Hashmi, A. S. K.; Toste, F. D., Eds.; Wiley: Weinheim, Germany, 2012; pp 237–262.
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] -
Liu, W.; Gust, R. Chem. Soc. Rev. 2013, 42, 755–773. doi:10.1039/C2CS35314H
Return to citation in text: [1] -
Chi, Y.; Chou, P.-T. Chem. Soc. Rev. 2010, 39, 638–655. doi:10.1039/B916237B
Return to citation in text: [1] -
Cheon, C. H.; Kanno, O.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 13248–13251. doi:10.1021/ja204331w
Return to citation in text: [1] -
Benitez, D.; Tkatchouk, E.; Gonzalez, A. Z.; Goddard, W. A., III; Toste, F. D. Org. Lett. 2009, 11, 4798–4801. doi:10.1021/ol9018002
Return to citation in text: [1] [2] -
Mauleón, P.; Zeldin, R. M.; González, A. Z.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 6348–6349. doi:10.1021/ja901649s
Return to citation in text: [1] [2] -
Hashmi, A. S. K.; Hengst, T.; Lothschütz, C.; Rominger, F. Adv. Synth. Catal. 2010, 352, 1315–1337. doi:10.1002/adsc.201000126
Return to citation in text: [1] [2] -
Kusama, H.; Karibe, Y.; Onizawa, Y.; Iwasawa, N. Angew. Chem., Int. Ed. 2010, 49, 4269–4272. doi:10.1002/anie.201001061
Return to citation in text: [1] [2] -
Jurberg, I. D.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 3543–3552. doi:10.1021/ja9100134
Return to citation in text: [1] [2] -
Bolte, B.; Gagosz, F. J. Am. Chem. Soc. 2011, 133, 7696–7699. doi:10.1021/ja202336p
Return to citation in text: [1] [2] -
Ye, L.; Wang, Y.; Aue, D. H.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 31–34. doi:10.1021/ja2091992
Return to citation in text: [1] [2] -
Hashmi, A. S. K.; Braun, I.; Nösel, P.; Schädlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Angew. Chem., Int. Ed. 2012, 51, 4456–4460. doi:10.1002/anie.201109183
Return to citation in text: [1] [2] -
Barluenga, J.; Sigüeiro, R.; Vicente, R.; Ballesteros, A.; Tomás, M.; Rodríguez, M. A. Angew. Chem., Int. Ed. 2012, 51, 10377–10381. doi:10.1002/anie.201205051
Return to citation in text: [1] [2] -
Mukherjee, P.; Widenhoefer, R. A. Chem.–Eur. J. 2013, 19, 3437–3444. doi:10.1002/chem.201203987
Return to citation in text: [1] [2] -
Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194
Return to citation in text: [1] [2] -
Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigüenza, J.; Díez, E.; Alonso, I.; Fernández, R.; Lassaletta, J. M.; López, F.; Mascareñas, J. L. J. Am. Chem. Soc. 2012, 134, 14322–14325. doi:10.1021/ja3065446
Return to citation in text: [1] [2] -
Yang, J.; Zhang, R.; Wang, W.; Zhang, Z.; Shi, M. Tetrahedron: Asymmetry 2011, 22, 2029–2038. doi:10.1016/j.tetasy.2011.12.004
Return to citation in text: [1] [2] -
Wang, W.; Yang, J.; Wang, F.; Shi, M. Organometallics 2011, 30, 3859–3869. doi:10.1021/om2004404
Return to citation in text: [1] [2] -
Liu, L.; Wang, F.; Wang, W.; Zhao, M.; Shi, M. Beilstein J. Org. Chem. 2011, 7, 555–564. doi:10.3762/bjoc.7.64
Return to citation in text: [1] [2] -
Wang, F.; Li, S.; Qu, M.; Zhao, M.-X.; Liu, L.-J.; Shi, M. Beilstein J. Org. Chem. 2012, 8, 726–731. doi:10.3762/bjoc.8.81
Return to citation in text: [1] [2] -
Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123
Return to citation in text: [1] [2] -
Shapiro, N. D.; Shi, Y.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 11654–11655. doi:10.1021/ja903863b
Return to citation in text: [1] [2] -
Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627
Return to citation in text: [1] [2] -
Teng, T.-M.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 9298–9300. doi:10.1021/ja1043837
Return to citation in text: [1] [2] -
Mukherjee, A.; Dateer, R. B.; Chaudhuri, R.; Bhunia, S.; Karad, S. N.; Liu, R.-S. J. Am. Chem. Soc. 2011, 133, 15372–15375. doi:10.1021/ja208150d
Return to citation in text: [1] [2] -
Wang, Y.-M.; Kuzniewski, C. N.; Rauniyar, V.; Hoong, C.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 12972–12975. doi:10.1021/ja205068j
Return to citation in text: [1] [2] -
Handa, S.; Slaughter, L. M. Angew. Chem., Int. Ed. 2012, 51, 2912–2915. doi:10.1002/anie.201107789
Return to citation in text: [1] [2] -
Pérez-Galán, P.; Delpont, N.; Herrero-Gómez, E.; Maseras, F.; Echavarren, A. M. Chem.–Eur. J. 2010, 16, 5324–5332. doi:10.1002/chem.200903507
Return to citation in text: [1] [2] -
Zhu, Z.-B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370
Return to citation in text: [1] [2] -
Lu, B.-L.; Wei, Y.; Shi, M. Chem.–Eur. J. 2010, 16, 10975–10979. doi:10.1002/chem.201001433
Return to citation in text: [1] [2] -
Jiang, M.; Liu, L.-P.; Shi, M.; Li, Y. Org. Lett. 2010, 12, 116–119. doi:10.1021/ol902593f
Return to citation in text: [1] [2] -
Zhang, D.-H.; Yao, L.-F.; Wei, Y.; Shi, M. Angew. Chem., Int. Ed. 2011, 50, 2583–2587. doi:10.1002/anie.201006969
Return to citation in text: [1] -
Huang, L.; Yang, H.-B.; Zhang, D.-H.; Zhang, Z.; Tang, X.-Y.; Xu, Q.; Shi, M. Angew. Chem., Int. Ed. 2013, 52, 6767–6771. doi:10.1002/anie.201302632
Return to citation in text: [1] -
Shi, M.; Liu, L.-P.; Tang, J. Org. Lett. 2006, 8, 4043–4046. doi:10.1021/ol0614830
Return to citation in text: [1] [2] -
Dai, L.-Z.; Qi, M.-J.; Shi, Y.-L.; Liu, X.-G.; Shi, M. Org. Lett. 2007, 9, 3191–3194. doi:10.1021/ol0713640
Return to citation in text: [1] [2] -
Tian, G.-Q.; Shi, M. Org. Lett. 2007, 9, 4917–4920. doi:10.1021/ol702341a
Return to citation in text: [1] [2] -
Dai, L.-Z.; Shi, M. Chem.–Eur. J. 2008, 14, 7011–7018. doi:10.1002/chem.200701954
Return to citation in text: [1] [2] -
Zhang, Z.; Shi, M. Chem.–Eur. J. 2010, 16, 7725–7729. doi:10.1002/chem.201000628
Return to citation in text: [1] [2] -
Lu, B.-L.; Shi, M. Chem.–Eur. J. 2011, 17, 9070–9075. doi:10.1002/chem.201100862
Return to citation in text: [1] [2] -
Lu, B.-L.; Dai, L.; Shi, M. Chem. Soc. Rev. 2012, 41, 3318–3339. doi:10.1039/C2CS15295A
Return to citation in text: [1] -
Nasiri, F.; Malakutikhah, D. Monatsh. Chem. 2011, 142, 807–812. doi:10.1007/s00706-011-0514-6
Return to citation in text: [1] -
Kabir, M. S.; Namjoshi, O. A.; Verma, R.; Lorenz, M.; Phani Babu Tiruveedhula, V. V. N.; Monte, A.; Bertz, S. H.; Schwabacher, A. W.; Cook, J. M. J. Org. Chem. 2012, 77, 300–310. doi:10.1021/jo201948e
Return to citation in text: [1] -
Tellam, J. P.; Kociok-Köhn, G.; Carbery, D. R. Org. Lett. 2008, 10, 5199–5202. doi:10.1021/ol802169j
Return to citation in text: [1] -
Fan, M.-J.; Li, G.-Q.; Li, L.-H.; Yang, S.-D.; Liang, Y.-M. Synthesis 2006, 2286–2292. doi:10.1055/s-2006-942437
Return to citation in text: [1] -
Mohrig, J. R.; Rosenberg, R. E.; Apostol, J. W.; Bastienaansen, M.; Evans, J. W.; Franklin, S. J.; Frisbie, C. D.; Fu, S. S.; Hamm, M. L.; Hirose, C. B.; Hunstad, D. A.; James, T. L.; King, R. W.; Larson, C. J.; Latham, H. A.; Owen, D. A.; Stein, K. A.; Warnet, R. J. Am. Chem. Soc. 1997, 119, 479–486. doi:10.1021/ja962631s
Return to citation in text: [1] -
Yavari, I.; Souri, S.; Sirouspour, M.; Djahaniani, H.; Nasiri, F. Synthesis 2005, 1761–1764. doi:10.1055/s-2005-865359
Return to citation in text: [1] -
Sarrafi, Y.; Sadatshahabi, M.; Alimohammadi, K.; Tajbakhsh, M. Green Chem. 2011, 13, 2851–2858. doi:10.1039/C1GC15625J
Return to citation in text: [1] -
Zhou, W.; Zhang, Y.; Li, P.; Wang, L. Org. Biomol. Chem. 2012, 10, 7184–7196. doi:10.1039/C2OB25969A
Return to citation in text: [1] -
Lautens, M.; Fagnou, K. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5455–5460. doi:10.1073/pnas.0307271101
Return to citation in text: [1]
| 1. | Tsui, G. C.; Tsoung, J.; Dougan, P.; Lautens, M. Org. Lett. 2012, 14, 5542–5545. doi:10.1021/ol302646a |
| 2. | Mannathan, S.; Cheng, C.-H. Chem. Commun. 2013, 49, 1557–1559. doi:10.1039/C2CC38001C |
| 3. | Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106–6108. doi:10.1039/C2CC31880F |
| 4. | Jack, K.; Fatila, E.; Hillis, C.; Tam, W. Synth. Commun. 2013, 43, 1181–1187. doi:10.1080/00397911.2011.626140 |
| 5. | Endo, K.; Tanaka, K.; Ogawa, M.; Shibata, T. Org. Lett. 2011, 13, 868–871. doi:10.1021/ol102928q |
| 6. | Madan, S.; Cheng, C.-H. J. Org. Chem. 2006, 71, 8312–8315. doi:10.1021/jo061477h |
| 7. | Lautens, M.; Rovis, T. J. Org. Chem. 1997, 62, 5246–5247. doi:10.1021/jo971115x |
| 8. | Barluenga, J.; Rodriguez, F.; Alvarez-Rodrigo, L.; Zapico, J. M.; Fañanás, F. J. Chem.–Eur. J. 2004, 10, 109–116. doi:10.1002/chem.200305374 |
| 22. | Bos, P. H.; Rudolph, A.; Pérez, M.; Fañanás-Mastral, M.; Harutyunyan, S. R.; Feringa, B. L. Chem. Commun. 2012, 48, 1748–1750. doi:10.1039/C2CC16855C |
| 23. | Sawama, Y.; Kawamoto, K.; Satake, H.; Krause, N.; Kita, Y. Synlett 2010, 2151–2155. doi:10.1055/s-0030-1258528 |
| 77. | Lautens, M.; Fagnou, K. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5455–5460. doi:10.1073/pnas.0307271101 |
| 16. | Tsui, G. C.; Dougan, P.; Lautens, M. Org. Lett. 2013, 15, 2652–2655. doi:10.1021/ol4009393 |
| 17. | Tsui, G. C.; Ninnemann, N. M.; Hosotani, A.; Lautens, M. Org. Lett. 2013, 15, 1064–1067. doi:10.1021/ol4000668 |
| 18. | Zhu, J.; Tsui, G. C.; Lautens, M. Angew. Chem., Int. Ed. 2012, 51, 12353–12356. doi:10.1002/anie.201207356 |
| 19. | Tsui, G. C.; Lautens, M. Angew. Chem., Int. Ed. 2012, 51, 5400–5404. doi:10.1002/anie.201200390 |
| 20. | Preetz, A.; Kohrt, C.; Drexler, H.-J.; Torrens, A.; Buschmann, H.; Lopez, M. G.; Heller, D. Adv. Synth. Catal. 2010, 352, 2073–2080. doi:10.1002/adsc.201000236 |
| 21. | Long, Y.; Zhao, S.; Zeng, H.; Yang, D. Catal. Lett. 2010, 138, 124–133. doi:10.1007/s10562-010-0383-3 |
| 11. | Hu, J.; Yang, Q.; Yu, L.; Xu, J.; Liu, S.; Huang, C.; Wang, L.; Zhou, Y.; Fan, B. Org. Biomol. Chem. 2013, 11, 2294–2301. doi:10.1039/C3OB27382B |
| 12. | Hu, J.; Yang, Q.; Xu, J.; Huang, C.; Fan, B.; Wang, J.; Lin, C.; Bian, Z.; Chan, A. S. C. Org. Biomol. Chem. 2013, 11, 814–820. doi:10.1039/C2OB26775F |
| 13. | Cheng, H.; Yang, D. J. Org. Chem. 2012, 77, 9756–9765. doi:10.1021/jo3018507 |
| 14. | Yang, D.; Long, Y.; Zhang, J.; Zeng, H.; Wang, S.; Li, C. Organometallics 2010, 29, 3477–3480. doi:10.1021/om100384q |
| 15. | Fan, B.-M.; Li, X.-J.; Peng, F.-Z.; Zhang, H.-B.; Chan, A. S. C.; Shao, Z.-H. Org. Lett. 2010, 12, 304–306. doi:10.1021/ol902574c |
| 57. | Zhu, Z.-B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 58. | Lu, B.-L.; Wei, Y.; Shi, M. Chem.–Eur. J. 2010, 16, 10975–10979. doi:10.1002/chem.201001433 |
| 59. | Jiang, M.; Liu, L.-P.; Shi, M.; Li, Y. Org. Lett. 2010, 12, 116–119. doi:10.1021/ol902593f |
| 62. | Shi, M.; Liu, L.-P.; Tang, J. Org. Lett. 2006, 8, 4043–4046. doi:10.1021/ol0614830 |
| 63. | Dai, L.-Z.; Qi, M.-J.; Shi, Y.-L.; Liu, X.-G.; Shi, M. Org. Lett. 2007, 9, 3191–3194. doi:10.1021/ol0713640 |
| 64. | Tian, G.-Q.; Shi, M. Org. Lett. 2007, 9, 4917–4920. doi:10.1021/ol702341a |
| 65. | Dai, L.-Z.; Shi, M. Chem.–Eur. J. 2008, 14, 7011–7018. doi:10.1002/chem.200701954 |
| 66. | Zhang, Z.; Shi, M. Chem.–Eur. J. 2010, 16, 7725–7729. doi:10.1002/chem.201000628 |
| 67. | Lu, B.-L.; Shi, M. Chem.–Eur. J. 2011, 17, 9070–9075. doi:10.1002/chem.201100862 |
| 68. | Lu, B.-L.; Dai, L.; Shi, M. Chem. Soc. Rev. 2012, 41, 3318–3339. doi:10.1039/C2CS15295A |
| 9. | Ge, G.-C.; Mo, D.-L.; Ding, C.-H.; Dai, L.-X.; Hou, X.-L. Org. Lett. 2012, 14, 5756–5759. doi:10.1021/ol302586m |
| 10. | Huang, X.-J.; Mo, D.-L.; Ding, C.-H.; Hou, X.-L. Synlett 2011, 943–946. doi:10.1055/s-0030-1259716 |
| 69. | Nasiri, F.; Malakutikhah, D. Monatsh. Chem. 2011, 142, 807–812. doi:10.1007/s00706-011-0514-6 |
| 70. | Kabir, M. S.; Namjoshi, O. A.; Verma, R.; Lorenz, M.; Phani Babu Tiruveedhula, V. V. N.; Monte, A.; Bertz, S. H.; Schwabacher, A. W.; Cook, J. M. J. Org. Chem. 2012, 77, 300–310. doi:10.1021/jo201948e |
| 71. | Tellam, J. P.; Kociok-Köhn, G.; Carbery, D. R. Org. Lett. 2008, 10, 5199–5202. doi:10.1021/ol802169j |
| 72. | Fan, M.-J.; Li, G.-Q.; Li, L.-H.; Yang, S.-D.; Liang, Y.-M. Synthesis 2006, 2286–2292. doi:10.1055/s-2006-942437 |
| 73. | Mohrig, J. R.; Rosenberg, R. E.; Apostol, J. W.; Bastienaansen, M.; Evans, J. W.; Franklin, S. J.; Frisbie, C. D.; Fu, S. S.; Hamm, M. L.; Hirose, C. B.; Hunstad, D. A.; James, T. L.; King, R. W.; Larson, C. J.; Latham, H. A.; Owen, D. A.; Stein, K. A.; Warnet, R. J. Am. Chem. Soc. 1997, 119, 479–486. doi:10.1021/ja962631s |
| 74. | Yavari, I.; Souri, S.; Sirouspour, M.; Djahaniani, H.; Nasiri, F. Synthesis 2005, 1761–1764. doi:10.1055/s-2005-865359 |
| 75. | Sarrafi, Y.; Sadatshahabi, M.; Alimohammadi, K.; Tajbakhsh, M. Green Chem. 2011, 13, 2851–2858. doi:10.1039/C1GC15625J |
| 76. | Zhou, W.; Zhang, Y.; Li, P.; Wang, L. Org. Biomol. Chem. 2012, 10, 7184–7196. doi:10.1039/C2OB25969A |
| 33. | Benitez, D.; Tkatchouk, E.; Gonzalez, A. Z.; Goddard, W. A., III; Toste, F. D. Org. Lett. 2009, 11, 4798–4801. doi:10.1021/ol9018002 |
| 34. | Mauleón, P.; Zeldin, R. M.; González, A. Z.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 6348–6349. doi:10.1021/ja901649s |
| 35. | Hashmi, A. S. K.; Hengst, T.; Lothschütz, C.; Rominger, F. Adv. Synth. Catal. 2010, 352, 1315–1337. doi:10.1002/adsc.201000126 |
| 36. | Kusama, H.; Karibe, Y.; Onizawa, Y.; Iwasawa, N. Angew. Chem., Int. Ed. 2010, 49, 4269–4272. doi:10.1002/anie.201001061 |
| 37. | Jurberg, I. D.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 3543–3552. doi:10.1021/ja9100134 |
| 38. | Bolte, B.; Gagosz, F. J. Am. Chem. Soc. 2011, 133, 7696–7699. doi:10.1021/ja202336p |
| 39. | Ye, L.; Wang, Y.; Aue, D. H.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 31–34. doi:10.1021/ja2091992 |
| 40. | Hashmi, A. S. K.; Braun, I.; Nösel, P.; Schädlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Angew. Chem., Int. Ed. 2012, 51, 4456–4460. doi:10.1002/anie.201109183 |
| 41. | Barluenga, J.; Sigüeiro, R.; Vicente, R.; Ballesteros, A.; Tomás, M.; Rodríguez, M. A. Angew. Chem., Int. Ed. 2012, 51, 10377–10381. doi:10.1002/anie.201205051 |
| 42. | Mukherjee, P.; Widenhoefer, R. A. Chem.–Eur. J. 2013, 19, 3437–3444. doi:10.1002/chem.201203987 |
| 43. | Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194 |
| 44. | Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigüenza, J.; Díez, E.; Alonso, I.; Fernández, R.; Lassaletta, J. M.; López, F.; Mascareñas, J. L. J. Am. Chem. Soc. 2012, 134, 14322–14325. doi:10.1021/ja3065446 |
| 45. | Yang, J.; Zhang, R.; Wang, W.; Zhang, Z.; Shi, M. Tetrahedron: Asymmetry 2011, 22, 2029–2038. doi:10.1016/j.tetasy.2011.12.004 |
| 46. | Wang, W.; Yang, J.; Wang, F.; Shi, M. Organometallics 2011, 30, 3859–3869. doi:10.1021/om2004404 |
| 47. | Liu, L.; Wang, F.; Wang, W.; Zhao, M.; Shi, M. Beilstein J. Org. Chem. 2011, 7, 555–564. doi:10.3762/bjoc.7.64 |
| 48. | Wang, F.; Li, S.; Qu, M.; Zhao, M.-X.; Liu, L.-J.; Shi, M. Beilstein J. Org. Chem. 2012, 8, 726–731. doi:10.3762/bjoc.8.81 |
| 57. | Zhu, Z.-B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 58. | Lu, B.-L.; Wei, Y.; Shi, M. Chem.–Eur. J. 2010, 16, 10975–10979. doi:10.1002/chem.201001433 |
| 59. | Jiang, M.; Liu, L.-P.; Shi, M.; Li, Y. Org. Lett. 2010, 12, 116–119. doi:10.1021/ol902593f |
| 60. | Zhang, D.-H.; Yao, L.-F.; Wei, Y.; Shi, M. Angew. Chem., Int. Ed. 2011, 50, 2583–2587. doi:10.1002/anie.201006969 |
| 61. | Huang, L.; Yang, H.-B.; Zhang, D.-H.; Zhang, Z.; Tang, X.-Y.; Xu, Q.; Shi, M. Angew. Chem., Int. Ed. 2013, 52, 6767–6771. doi:10.1002/anie.201302632 |
| 25. | Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/C1CS15088J |
| 26. | Sethofer, S. G.; Mayer, T.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8276–8277. doi:10.1021/ja103544p |
| 27. | Rudolph, M. In Modern Gold Catalyzed Synthesis; Hashmi, A. S. K.; Toste, F. D., Eds.; Wiley: Weinheim, Germany, 2012; pp 331–362. |
| 28. | Hubbert, C.; Hashmi, A. S. K. In Modern Gold Catalyzed Synthesis; Hashmi, A. S. K.; Toste, F. D., Eds.; Wiley: Weinheim, Germany, 2012; pp 237–262. |
| 29. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 30. | Liu, W.; Gust, R. Chem. Soc. Rev. 2013, 42, 755–773. doi:10.1039/C2CS35314H |
| 31. | Chi, Y.; Chou, P.-T. Chem. Soc. Rev. 2010, 39, 638–655. doi:10.1039/B916237B |
| 32. | Cheon, C. H.; Kanno, O.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 13248–13251. doi:10.1021/ja204331w |
| 33. | Benitez, D.; Tkatchouk, E.; Gonzalez, A. Z.; Goddard, W. A., III; Toste, F. D. Org. Lett. 2009, 11, 4798–4801. doi:10.1021/ol9018002 |
| 34. | Mauleón, P.; Zeldin, R. M.; González, A. Z.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 6348–6349. doi:10.1021/ja901649s |
| 35. | Hashmi, A. S. K.; Hengst, T.; Lothschütz, C.; Rominger, F. Adv. Synth. Catal. 2010, 352, 1315–1337. doi:10.1002/adsc.201000126 |
| 36. | Kusama, H.; Karibe, Y.; Onizawa, Y.; Iwasawa, N. Angew. Chem., Int. Ed. 2010, 49, 4269–4272. doi:10.1002/anie.201001061 |
| 37. | Jurberg, I. D.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 3543–3552. doi:10.1021/ja9100134 |
| 38. | Bolte, B.; Gagosz, F. J. Am. Chem. Soc. 2011, 133, 7696–7699. doi:10.1021/ja202336p |
| 39. | Ye, L.; Wang, Y.; Aue, D. H.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 31–34. doi:10.1021/ja2091992 |
| 40. | Hashmi, A. S. K.; Braun, I.; Nösel, P.; Schädlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Angew. Chem., Int. Ed. 2012, 51, 4456–4460. doi:10.1002/anie.201109183 |
| 41. | Barluenga, J.; Sigüeiro, R.; Vicente, R.; Ballesteros, A.; Tomás, M.; Rodríguez, M. A. Angew. Chem., Int. Ed. 2012, 51, 10377–10381. doi:10.1002/anie.201205051 |
| 42. | Mukherjee, P.; Widenhoefer, R. A. Chem.–Eur. J. 2013, 19, 3437–3444. doi:10.1002/chem.201203987 |
| 43. | Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194 |
| 44. | Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigüenza, J.; Díez, E.; Alonso, I.; Fernández, R.; Lassaletta, J. M.; López, F.; Mascareñas, J. L. J. Am. Chem. Soc. 2012, 134, 14322–14325. doi:10.1021/ja3065446 |
| 45. | Yang, J.; Zhang, R.; Wang, W.; Zhang, Z.; Shi, M. Tetrahedron: Asymmetry 2011, 22, 2029–2038. doi:10.1016/j.tetasy.2011.12.004 |
| 46. | Wang, W.; Yang, J.; Wang, F.; Shi, M. Organometallics 2011, 30, 3859–3869. doi:10.1021/om2004404 |
| 47. | Liu, L.; Wang, F.; Wang, W.; Zhao, M.; Shi, M. Beilstein J. Org. Chem. 2011, 7, 555–564. doi:10.3762/bjoc.7.64 |
| 48. | Wang, F.; Li, S.; Qu, M.; Zhao, M.-X.; Liu, L.-J.; Shi, M. Beilstein J. Org. Chem. 2012, 8, 726–731. doi:10.3762/bjoc.8.81 |
| 49. | Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 50. | Shapiro, N. D.; Shi, Y.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 11654–11655. doi:10.1021/ja903863b |
| 51. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 52. | Teng, T.-M.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 9298–9300. doi:10.1021/ja1043837 |
| 53. | Mukherjee, A.; Dateer, R. B.; Chaudhuri, R.; Bhunia, S.; Karad, S. N.; Liu, R.-S. J. Am. Chem. Soc. 2011, 133, 15372–15375. doi:10.1021/ja208150d |
| 54. | Wang, Y.-M.; Kuzniewski, C. N.; Rauniyar, V.; Hoong, C.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 12972–12975. doi:10.1021/ja205068j |
| 55. | Handa, S.; Slaughter, L. M. Angew. Chem., Int. Ed. 2012, 51, 2912–2915. doi:10.1002/anie.201107789 |
| 56. | Pérez-Galán, P.; Delpont, N.; Herrero-Gómez, E.; Maseras, F.; Echavarren, A. M. Chem.–Eur. J. 2010, 16, 5324–5332. doi:10.1002/chem.200903507 |
| 62. | Shi, M.; Liu, L.-P.; Tang, J. Org. Lett. 2006, 8, 4043–4046. doi:10.1021/ol0614830 |
| 63. | Dai, L.-Z.; Qi, M.-J.; Shi, Y.-L.; Liu, X.-G.; Shi, M. Org. Lett. 2007, 9, 3191–3194. doi:10.1021/ol0713640 |
| 64. | Tian, G.-Q.; Shi, M. Org. Lett. 2007, 9, 4917–4920. doi:10.1021/ol702341a |
| 65. | Dai, L.-Z.; Shi, M. Chem.–Eur. J. 2008, 14, 7011–7018. doi:10.1002/chem.200701954 |
| 66. | Zhang, Z.; Shi, M. Chem.–Eur. J. 2010, 16, 7725–7729. doi:10.1002/chem.201000628 |
| 67. | Lu, B.-L.; Shi, M. Chem.–Eur. J. 2011, 17, 9070–9075. doi:10.1002/chem.201100862 |
| 24. | Brazeau, J.-F.; Zhang, S.; Colomer, I.; Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2012, 134, 2742–2749. doi:10.1021/ja210388g |
| 23. | Sawama, Y.; Kawamoto, K.; Satake, H.; Krause, N.; Kita, Y. Synlett 2010, 2151–2155. doi:10.1055/s-0030-1258528 |
| 49. | Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 50. | Shapiro, N. D.; Shi, Y.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 11654–11655. doi:10.1021/ja903863b |
| 51. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 52. | Teng, T.-M.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 9298–9300. doi:10.1021/ja1043837 |
| 53. | Mukherjee, A.; Dateer, R. B.; Chaudhuri, R.; Bhunia, S.; Karad, S. N.; Liu, R.-S. J. Am. Chem. Soc. 2011, 133, 15372–15375. doi:10.1021/ja208150d |
| 54. | Wang, Y.-M.; Kuzniewski, C. N.; Rauniyar, V.; Hoong, C.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 12972–12975. doi:10.1021/ja205068j |
| 55. | Handa, S.; Slaughter, L. M. Angew. Chem., Int. Ed. 2012, 51, 2912–2915. doi:10.1002/anie.201107789 |
| 56. | Pérez-Galán, P.; Delpont, N.; Herrero-Gómez, E.; Maseras, F.; Echavarren, A. M. Chem.–Eur. J. 2010, 16, 5324–5332. doi:10.1002/chem.200903507 |
© 2013 Sun et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)