Abstract
A combined experimental and computational study on regioselective gold-catalyzed synthetic routes to 1,3-oxazinan-2-ones (kinetically controlled products) and 1,3-oxazin-2-one derivatives (thermodynamically favored) from easily accessible allenic carbamates has been carried out.

Graphical Abstract
Introduction
The search for new synthetic routes to 1,3-oxazin-2-one derivatives [1] is of interest because of the biological activity of these molecules [2-7]. Carretero and colleagues have published the Au(I)-catalyzed cyclization of N-Boc-3-butyn-1-amines to afford six-membered 2-oxazinones, namely, 6-methylene-1,3-oxazinan-2-ones involving a 6-exo-dig cyclization [1]. This interesting method avoids complicated prefunctionalization of starting materials and minimizes the formation of byproducts; however, it is used just for the preparation of four simple examples. 1,3-Oxazin-2-ones are also used as valuable intermediates in organic synthesis [8-14]. Recently, allenes have attracted much attention as they have been used for the preparation of both biologically relevant drugs as well as advanced materials [15-27]. However, regioselectivity problems are significant (endo-trig versus endo-dig versus exo-dig versus exo-trig cyclization). Great effort is currently being made in the search for a variety of reactions promoted by gold salts due to their impressive catalytic properties [28-41]. The Boc protective group has been widely used in allene chemistry, being an inert and recommended partner in gold- and palladium-catalyzed aminocyclizations of allenes [42]. On the other hand, reports of gold-catalyzed cyclizations leading to heterocycles that contain more than one heteroatom are rare [43-48]. Besides, it has been reported very recently that the Au(I)-catalyzed cyclization of a N-phenethyl-N-Boc-protected allenamide failed [49]. Despite the above precedents, but in continuation of our interest in heterocyclic and allene chemistry [50-55], we decided to examine the gold-catalyzed cyclization of N-Boc-allenes with the aim of establishing a protocol for the synthesis of 1,3-oxazin-2-one derivatives in which the carbamate group should serve as the source of CO2.
Results and Discussion
To explore the effects of various substrates on gold-catalyzed oxycyclization reactions, a number of new allenic carbamates were synthesized as shown in Scheme 1. Starting materials, tert-butyl (prop-2-ynyl)carbamates 1a–j, were obtained both in the racemic form and in optically pure form by using standard methodologies. Thus, alkynylcarbamates 1a–g were prepared through reductive amination of the appropriate aldehyde with propargylamine, followed by Boc2O treatment of the corresponding N-substituted prop-2-yn-1-amine. Alkynylcarbamate 1h was prepared from Garner’s aldehyde following a literature report [56,57]. Alkynylcarbamate 1i was readily accessed from (S)-prolinol by using a modified known procedure [58]. Alkynylcarbamate 1j was achieved through the reaction of 3-bromo-1H-indole-2-carbaldehyde with the Ohira–Bestmann reagent followed by the addition of Boc2O. Terminal alkynes 1 were conveniently converted into allenic carbamates 2 by treatment with paraformaldehyde in the presence of diisopropylamine and copper(I) bromide (Crabbé reaction) [59,60].
![[1860-5397-9-93-i1]](/bjoc/content/inline/1860-5397-9-93-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of allenic carbamates 2a–j. Reagents and conditions: (i) Propargylamine, MgSO4, CH2Cl2, rt, 15 h. (ii) NaBH4, MeOH, rt, 0.5 h. (iii) Boc2O, Et3N, CH2Cl2, rt, 2–15 h. (iv) (CH2O)n, iPr2NH, CuBr, 1,4-dioxane, reflux, 1 h. (v) Ohira–Bestmann reagent, K2CO3, MeOH, rt, 15 h. (vi) Boc2O, DMAP, CH3CN, rt, 2 h. (vii) Dess–Martin periodinane, CH2Cl2, rt. PMP = 4-MeOC6H4.
Scheme 1: Preparation of allenic carbamates 2a–j. Reagents and conditions: (i) Propargylamine, MgSO4, CH2Cl2,...
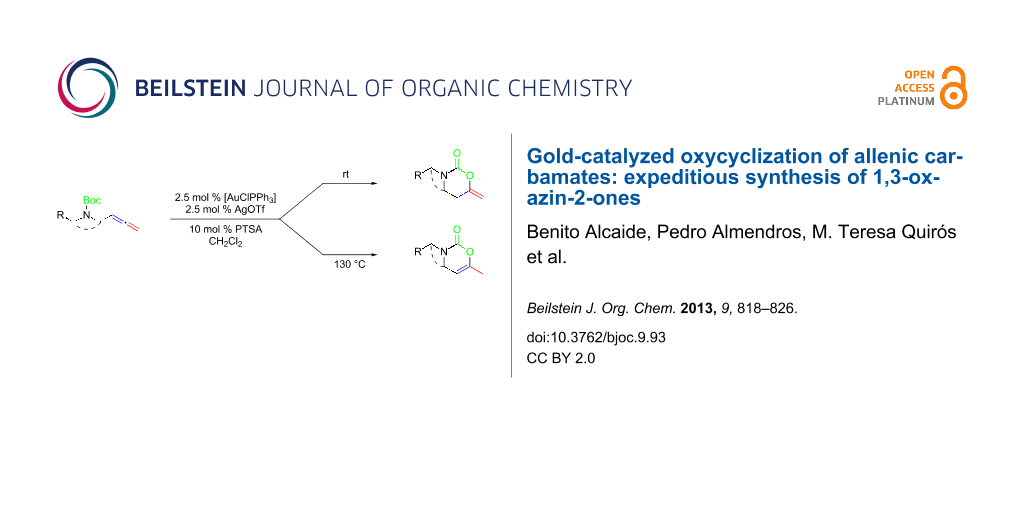
We employed three different gold salts in our initial screening of catalysts for the model system, allenic carbamate 2a. Initially, the use of AuCl3 and AuCl were tested, but both failed to catalyze the reaction. Fortunately, we found that [AuClPPh3]/AgOTf was an excellent catalyst for our purpose. To our delight, the reaction of allenic carbamate 2a at room temperature afforded 3-benzyl-6-methylene-1,3-oxazinan-2-one (3a) bearing an exocyclic double bond as the sole product (Scheme 2). Adding a catalytic amount of Brønsted acid (PTSA) into the reaction system did slightly improve the yield of 3a. Solvent screening demonstrated that dichloromethane was the best choice in the reaction.
![[1860-5397-9-93-i2]](/bjoc/content/inline/1860-5397-9-93-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Controlled oxycyclization reactions of allenic carbamates 2 to 1,3-oxazinan-2-ones 3 and 1,3-oxazin-2-ones 4 under selective gold-catalyzed conditions. Reagents and conditions: (i) 2.5 mol % [AuClPPh3], 2.5 mol % AgOTf, 10 mol % PTSA, CH2Cl2, rt, 3a: 6 h; 3b: 7 h, 3d: 6 h; 3e: 8 h; 3f: 7 h; 3g: 5.5 h; 3h: 5 h; 3i: 7 h; 3j: 2 h. (ii) 2.5 mol % [AuClPPh3], 2.5 mol % AgOTf, 10 mol % PTSA, CH2Cl2, sealed tube, 130 °C, 4a: 1.5 h; 4b: 2 h, 4c: 4 h; 4d: 6 h; 4e: 4 h; 4f: 0.5 h; 4g: 5.5 h; 4i: 2 h. (iii) 2.5 mol % [AuClPPh3], 2.5 mol % AgOTf, 10 mol % PTSA, CH2Cl2, sealed tube, 80 °C, 4j: 2 h. PMP = 4-MeOC6H4.
Scheme 2: Controlled oxycyclization reactions of allenic carbamates 2 to 1,3-oxazinan-2-ones 3 and 1,3-oxazin...
As revealed in Scheme 2, a variety of allenic carbamates 2 were also suitable for such heterocyclization reactions to afford 1,3-oxazinan-2-ones 3. To increase the molecular diversity by incorporating more 1,3-oxazin-2-ones in the molecule, compound 2g having two allenic carbamate units was used. Notably, bis(allenic carbamate) 2g also undergoes this interesting transformation to give bis(6-methylene-1,3-oxazinan-2-one) 3g through a two-fold cyclization. This product particularly underlines the power of the present cyclization reaction, as none of the conventional methods would allow its synthesis with such great ease.
Interestingly, as a first try, we were pleased to notice that the reaction of allenyl derivative 2a in dichloromethane at 90 °C, afforded 1,3-oxazin-2-one 4a bearing an endocyclic double bond as the major component, and 1,3-oxazinan-2-one 3a was also isolated as a minor component. Notably, starting from allenic carbamates 2a–j and performing the reaction in dichloromethane at 130 °C, a series of 6-methyl-3-substituted 3,4-dihydro-2H-1,3-oxazin-2-ones 4a–j were exclusively formed (Scheme 2) [61-68]. The observed regioselectivity is worthy of note, because under our reaction conditions only 1,3-oxazinan-2-ones 3 (arising from 6-endo-dig cyclization) or 3,4-dihydro-2H-1,3-oxazin-2-ones 4 (arising from 6-exo-dig cyclization) were achieved, with the nucleophilic oxygen attacking the central allene carbon atom in each case. This is an interesting result, because the available examples on related metal-catalyzed allene heterocyclizations usually lead to 5-exo-trig cyclization [69,70]; only Hashmi et al. have recently reported an attack at the central position of the allene in allenylamides [44].
Thus, it is possible to suppress the formation of the 1,3-oxazinan-2-one ring by performing the reaction at higher temperature, yielding the 1,3-oxazin-2-one as the exclusive product. A general trend can be deduced on the basis of these results: heterocycle 4 is the thermodynamically controlled product while heterocycle 3 is the kinetically controlled product [71-73]. Probably, double-bond migration in compounds 3 results in the formation of the 1,3-oxazin-2-one 4. In order to verify the role of the Au(I) catalyst in the double-bond migration process, we set up two experiments. Heating a mixture of 3a with Au(OTf)PPh3 at a loading of 2.5 mol % in dichloromethane for 1.5 h at 130 °C resulted in full conversion into 4a. Running the same reaction in the absence of any catalyst resulted in 30% conversion after two days, as determined by 1H NMR. Treatment of 1,3-oxazinan-2-one 3a with 5 mol % TfOH in CH2Cl2 at room temperature did not proceed to give an appreciable amount of 3,4-dihydro-2H-1,3-oxazin-2-one 4a after 2 h. This indicates that the Au(I) catalyst might participate in the double-bond migration process; being a possible intermediate, the π-allyl complex 5 is depicted in Scheme 3 [74]. Despite that, the isomerization process can be also viewed as an intramolecular 1,3-H shift assisted by gold.
![[1860-5397-9-93-i3]](/bjoc/content/inline/1860-5397-9-93-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible explanation for the gold-catalyzed isomerization reaction of 1,3-oxazinan-2-one 3a into 1,3-oxazin-2-one 4a. Reagents and conditions: (i) 2.5 mol % [AuClPPh3], 2.5 mol % AgOTf, 10 mol % PTSA, CH2Cl2, sealed tube, 130 °C, 1.5 h.
Scheme 3: Possible explanation for the gold-catalyzed isomerization reaction of 1,3-oxazinan-2-one 3a into 1,...
A possible pathway for the gold-catalyzed achievement of heterocycles 3 from allenyl-tethered carbamates 2 may initially involve the formation of a complex 6 through coordination of the gold salt to the proximal allenic double bond. Next, chemo- and regioselective 6-endo-dig oxyauration of the carbamate carbonyl moiety forms species 7. Attack of the carbamate carbonyl group occurs as a result of the stability of the intermediate ammonium cation type 7. Loss of proton linked to 2-methylprop-1-ene release [75-78], generates neutral species 8, which followed by protonolysis of the carbon–gold bond affords 6-methylene-1,3-oxazinan-2-ones 3 with concurrent regeneration of the gold catalyst (Scheme 4, left catalytic cycle). In line with the above mechanistic proposal, the easy breakage of the tert-butyl group at species 7 is essential for the formation of 1,3-oxazinan-2-ones 3. Besides, the replacement of the tert-butyl group in allenic carbamates 2 by other alkyl functions, such as methyl, did not allow the preparation of heterocycles 3. In addition to the double-bond isomerization that transforms products 3 into the thermodynamically more favored compounds 4, a mechanistic scenario involving the initial coordination of the gold to the distal allenic double bond leading to complex 9, followed by a 6-exo-dig oxyauration is likely for the achievement of 1,3-oxazin-2-ones 4 from allenic carbamates 2 (Scheme 4, right-hand catalytic cycle).
![[1860-5397-9-93-i4]](/bjoc/content/inline/1860-5397-9-93-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanistic explanation for the gold-catalyzed oxycyclization reactions of allenic carbamates 2 into 6-methylene-1,3-oxazinan-2-ones 3 or into 3-substituted-3,4-dihydro-2H-1,3-oxazin-2-ones 4.
Scheme 4: Mechanistic explanation for the gold-catalyzed oxycyclization reactions of allenic carbamates 2 int...
Density functional theory (DFT) calculations (see Supporting Information File 1) have been carried out at the PCM-M06/def2-SVP//B3LYP/def2-SVP level to gain more insight into the reaction mechanism of the above discussed gold-catalyzed divergent oxycyclization reaction. The corresponding computed reaction profiles of the model allene 1M with the model gold catalyst AuPMe3(OTf) are shown in Figure 1, which gathers the respective free energies (computed at 298 K) in CH2Cl2 solution.
![[1860-5397-9-93-1]](/bjoc/content/figures/1860-5397-9-93-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Computed reaction profile for the reaction of AuPMe3+ and 1M. Numbers indicate the corresponding PCM-corrected ΔG298 energies (in kcal/mol) using dichloromethane as solvent. Bond distances are given in angstroms. All data have been computed at the PCM-M06/def2-SVP//B3LYP/def2-SVP level.
Figure 1: Computed reaction profile for the reaction of AuPMe3+ and 1M. Numbers indicate the corresponding PC...
As initially envisaged, two different coordination modes of the metal fragment to the allenic double bond of 1M, i.e., distal versus proximal, are possible. Our calculations indicate that the distal coordination leading to INT1-B is favored over the proximal coordination mode, which forms INT1-A (ΔΔG = 3.8 kcal/mol). This is mainly due to the presence of a two-electron stabilizing interaction established by donation of electronic density from the lone pair of the oxygen atom of the carbonyl moiety to a vacant d atomic orbital of the gold atom in INT1-B [79]. Both complexes can undergo the corresponding oxyauration cyclization reaction. Thus, INT1-A is converted into INT2-A in a slightly exergonic process (ΔGR,298 = −0.4 kcal/mol) through the saddle point TS1-A, which is associated with the 6-endo-dig cyclization. Similarly, INT1-B is transformed into INT2-B in a slightly endergonic process (ΔGR,298 = +0.9 kcal/mol) via TS1-B, associated with the 6-exo-dig cyclization reaction.
From the data in Figure 1, it becomes obvious that the 6-endo-dig transformation is kinetically favored over the 6-exo-dig reaction in view of the computed lower activation barrier of the former process (ΔΔG≠298 = +7.4 kcal/mol). However, the cyclic reaction product INT2-B is thermodynamically more stable than the counterpart INT2-A (ΔΔG = 2.5 kcal/mol), which is in agreement with the experimental findings (see above). The next step of the process involves the TfO− promoted elimination of isobutene to form the corresponding INT3 complexes. The driving force of this process is clearly related to the thermodynamically favored release of isobutene (ΔGR,298 = −1.9 and −5.2 kcal/mol from INT3-A and INT3-B, respectively). Finally, the protonolysis reaction of the carbon–gold bond by TfOH renders the final products 3M and 4M regenerating the catalyst. This step occurs through the transition states TS2-A and TS2-B, respectively, in an exergonic transformation (ΔGR,298 = −4.5 and −3.0 kcal/mol from INT3-A and INT3-B, respectively). Again, the data in Figure 1 indicate that the final product 4M is thermodynamically more stable than 3M, which is in line with the experimentally observed conversion of 3a into 4a by heating in the presence and also in the absence of the gold-catalyst. From the computed reaction profile, it can be concluded that the observed divergent cyclization finds its origin in the initial 6-endo versus 6-exo oxyauration reaction steps, with the former being kinetically favored whereas the latter is thermodynamically favored. At this point, it cannot be safely discarded that the formation of the thermodynamically more stable 6-exo-dig products is the result of the simple thermally promoted isomerization of the less stable 6-endo-dig species.
Conclusion
In conclusion, efficient gold-catalyzed synthetic routes to 1,3-oxazinan-2-one and 1,3-oxazin-2-one derivatives from easily accessible allenic carbamates under mild conditions have been reported. The oxycyclization reactions were found to proceed with complete control of regioselectivity. The mechanism of these processes has additionally been investigated by a computational study showing that heterocycles 3 are the kinetically controlled products whereas heterocycles 4 are thermodynamically favored.
Experimental
General Information
1H NMR and 13C NMR spectra were recorded on 700, 500, 300, or 200 MHz spectrometers. NMR spectra were recorded in CDCl3 solutions, except were otherwise stated. Chemical shifts are given in parts per million relative to TMS (1H, 0.0 ppm) or CDCl3 (13C, 76.9 ppm). Low- and high-resolution mass spectra were taken on a QTOF LC–MS spectrometer using the electronic impact (EI) or electrospray modes (ES) unless otherwise stated. Specific rotation [α]D is given in 10−1 deg cm2 g−1 at 20 °C, and the concentration (c) is expressed in grams per 100 mL. All commercially available compounds were used without further purification.
Typical procedure for the Au(I)-catalyzed preparation of 1,3-oxazin-2-ones, 4
[AuClPPh3] (0.00475 mmol), AgOTf (0.00475 mmol), and p-toluenesulfonic acid (0.019 mmol) were sequentially added to a stirred solution of the allenic carbamate 2a (50 mg, 0.19 mmol) in dichloromethane (1.9 mL). The resulting mixture was heated in a sealed tube at 130 °C until disappearance of the starting material (TLC, 1.5 h). The reaction was allowed to cool to room temperature and filtered through a pack of celite. The filtrate was extracted with dichloromethane (3 × 5 mL), and the combined extracts were washed twice with brine. The organic layer was dried (MgSO4), concentrated under reduced pressure, and purified by flash column chromatography on silica gel (hexanes/ethyl acetate 4:1) to afford product 4a (27 mg, 70%) as a colorless oil. 1H NMR (300 MHz, CDCl3, 25 °C) δ 7.33 (m, 2H), 4.76 (m, 1H), 4.58 (s, 2H), 3.68 (dq, J = 3.2, 1.9 Hz, 2H), 1.86 (td, J = 1.9, 1.2 Hz, 3H); 13C NMR (75 MHz, CDCl3, 25 °C) δ 150.9, 148.1, 135.6, 128.7, 128.2, 127.9, 94.4, 52.1, 44.6, 18.3; IR (CHCl3) ν: 1685 cm−1; HRMS–ES (m/z): [M]+ calcd for C12H13NO2, 203.0946; found, 203.0952.
Supporting Information
| Supporting Information File 1: Experimental details, analytical data of new compounds, copies of 1H NMR and 13C NMR spectra and computational details. | ||
| Format: PDF | Size: 2.1 MB | Download |
Acknowledgements
Financial support from the MINECO [Projects CTQ2012-33664-C02-01, CTQ2012-33664-C02-02, CTQ2010-20714-C02-01, and Consolider-Ingenio 2010 (CSD2007-00006)] and Comunidad Autónoma de Madrid (Projects S2009/PPQ-1752 and S2009/PPQ-1634) are gratefully acknowledged. M. T. Q. thanks MEC for a predoctoral grant.
References
-
Robles-Machín, R.; Adrio, J.; Carretero, J. C. J. Org. Chem. 2006, 71, 5023. doi:10.1021/jo060520y
See for the synthesis of oxazinones from N-Boc-(3-butyn)-1-amines.
Return to citation in text: [1] [2] -
Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem., Int. Ed. 2012, 51, 752. doi:10.1002/anie.201106249
Return to citation in text: [1] -
Wolfe, A. L.; Duncan, K. K.; Parelkar, N. K.; Weir, S. J.; Vielhauer, G. A.; Boger, D. L. J. Med. Chem. 2012, 55, 5878. doi:10.1021/jm300330b
Return to citation in text: [1] -
Taft, F.; Harmrolfs, K.; Nickeleit, I.; Heutling, A.; Kiene, M.; Malek, N.; Sasse, F.; Kirschning, A. Chem.–Eur. J. 2012, 18, 880. doi:10.1002/chem.201101640
Return to citation in text: [1] -
Chinkov, N.; Warm, A.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 2957. doi:10.1002/anie.201006689
Return to citation in text: [1] -
Fishkin, N.; Maloney, E. K.; Chari, R. V. J.; Singh, R. Chem. Commun. 2011, 47, 10752. doi:10.1039/c1cc14164c
Return to citation in text: [1] -
Zhang, F.; Qian, L.; Flood, P. M.; Shi, J.-S.; Hong, J.-S.; Gao, H.-M. J. Pharmacol. Exp. Ther. 2010, 333, 822. doi:10.1124/jpet.110.165829
Return to citation in text: [1] -
Richter, H.; Fröhlich, R.; Daniliuc, C.-G.; García-Mancheño, O. Angew. Chem., Int. Ed. 2012, 51, 8656. doi:10.1002/anie.201202379
Return to citation in text: [1] -
Kuznetsov, N. Yu.; Maleev, V. I.; Khrustalev, V. N.; Mkrtchyan, A. F.; Godovikov, I. A.; Strelkova, T. V.; Bubnov, Y. N. Eur. J. Org. Chem. 2012, 334. doi:10.1002/ejoc.201101114
Return to citation in text: [1] -
Shpak-Kraievskyi, P.; Yin, B.; Martel, A.; Dhal, R.; Dujardin, G.; Laurent, M. Y. Tetrahedron 2012, 68, 2179. doi:10.1016/j.tet.2012.01.002
Return to citation in text: [1] -
Tian, C.; Jiao, X.; Liu, X.; Li, R.; Dong, L.; Liu, X.; Zhang, Z.; Xu, J.; Xu, M.; Xie, P. Tetrahedron Lett. 2012, 53, 4892. doi:10.1016/j.tetlet.2012.07.011
Return to citation in text: [1] -
Zhou, H.-B.; Lee, J. H.; Mayne, C. G.; Carlson, K. E.; Katzenellenbogen, J. A. J. Med. Chem. 2010, 53, 3349. doi:10.1021/jm100052k
Return to citation in text: [1] -
Sato, S.; Shibuya, M.; Kanoh, N.; Iwabuchi, Y. Chem. Commun. 2009, 6264. doi:10.1039/b913770j
Return to citation in text: [1] -
Osa, Y.; Hikima, Y.; Sato, Y.; Takino, K.; Ida, Y.; Hirono, S.; Nagase, H. J. Org. Chem. 2005, 70, 5737. doi:10.1021/jo0501644
Return to citation in text: [1] -
Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074. doi:10.1002/anie.201101460
Return to citation in text: [1] -
Rivera-Fuentes, P.; Diederich, F. Angew. Chem., Int. Ed. 2012, 51, 2818. doi:10.1002/anie.201108001
Return to citation in text: [1] -
Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994. doi:10.1021/cr1004088
Return to citation in text: [1] -
Alcaide, B.; Almendros, P. Chem. Rec. 2011, 11, 311. doi:10.1002/tcr.201100011
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Soc. Rev. 2010, 39, 783. doi:10.1039/b913749a
Return to citation in text: [1] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45. doi:10.1021/ar800011h
Return to citation in text: [1] -
Krause, N., Ed. Compounds with all carbon functions: Cumulenes and Allenes; Science of Synthesis; Georg Thieme: Stuttgart, Germany, 2007; Vol. 44.
Return to citation in text: [1] -
Ma, S. Chem. Rev. 2005, 105, 2829. doi:10.1021/cr020024j
Return to citation in text: [1] -
Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004.
Return to citation in text: [1] -
Alcaide, B.; Almendros, P. Eur. J. Org. Chem. 2004, 3377. doi:10.1002/ejoc.200400023
Return to citation in text: [1] -
Bates, R. W.; Satcharoen, V. Chem. Soc. Rev. 2002, 31, 12. doi:10.1039/b103904k
Return to citation in text: [1] -
Zimmer, R.; Dinesh, C. U.; Nandanan, E.; Khan, F. A. Chem. Rev. 2000, 100, 3067. doi:10.1021/cr9902796
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem., Int. Ed. 2000, 39, 3590. doi:10.1002/1521-3773(20001016)39:20<3590::AID-ANIE3590>3.0.CO;2-L
Return to citation in text: [1] -
Hashmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012.
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657. doi:10.1021/cr100414u
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536. doi:10.1039/c1cc10780a
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Alonso, J. M. Org. Biomol. Chem. 2011, 9, 4405. doi:10.1039/c1ob05249g
Return to citation in text: [1] -
Bandini, M. Chem. Soc. Rev. 2011, 40, 1358. doi:10.1039/c0cs00041h
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078
Return to citation in text: [1] -
Lipshutz, B.; Yamamoto, Y., Eds. Coinage Metals in Organic Synthesis. Chem. Rev. 2008, 108, 2793–3442.
Return to citation in text: [1] -
Hutchings, G. J.; Brust, M.; Schmidbaur, H., Eds. Gold – Chemistry, Materials and Catalysis. Chem. Soc. Rev. 2008, 37, 1745–2140.
Return to citation in text: [1] -
Muzart, J. Tetrahedron 2008, 64, 5815. doi:10.1016/j.tet.2008.04.018
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333. doi:10.1039/b612008c
Return to citation in text: [1] -
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271. doi:10.1002/adsc.200600368
See for a review.
Return to citation in text: [1] -
Alcaide, B.; Almendros, P. Adv. Synth. Catal. 2011, 353, 2561. doi:10.1002/adsc.201100160
Return to citation in text: [1] -
Higginbotham, M. C. M.; Bebbington, M. W. P. Chem. Commun. 2012, 48, 7565. doi:10.1039/c2cc33711h
Return to citation in text: [1] -
Hashmi, A. S. K.; Schuster, A. M.; Litters, S.; Rominger, F.; Pernpointner, M. Chem.–Eur. J. 2011, 17, 5661. doi:10.1002/chem.201100132
Return to citation in text: [1] [2] -
LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598. doi:10.1002/anie.200905000
Return to citation in text: [1] -
Li, H.; Widenhoefer, R. A. Org. Lett. 2009, 11, 2671. doi:10.1021/ol900730w
Return to citation in text: [1] -
Winter, C.; Krause, N. Angew. Chem., Int. Ed. 2009, 48, 6339. doi:10.1002/anie.200902355
Return to citation in text: [1] -
Yeom, H.-S.; Lee, E.-S.; Shin, S. Synlett 2007, 2292. doi:10.1055/s-2007-985571
Return to citation in text: [1] -
Singh, S.; Elsegood, M. R. J.; Kimber, M. C. Synlett 2012, 565. doi:10.1055/s-0031-1290335
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Quirós, M. T.; López, R.; Menéndez, M. I.; Sochacka-Ćwikła, A. J. Am. Chem. Soc. 2013, 135, 898. doi:10.1021/ja3108966
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Cembellín, S.; Martínez del Campo, T.; Fernández, I. Chem. Commun. 2013, 49, 1282. doi:10.1039/c2cc37872h
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Alonso, J. M.; Fernández, I. Chem. Commun. 2012, 48, 6604. doi:10.1039/c2cc32015k
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Aragoncillo, C.; Gómez-Campillos, G.; Arnó, M.; Domingo, L. R. ChemPlusChem 2012, 77, 563. doi:10.1002/cplu.201200090
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Alonso, J. M.; Quirós, M. T.; Gadziński, P. Adv. Synth. Catal. 2011, 353, 1871. doi:10.1002/adsc.201100209
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Carrascosa, R. Chem.–Eur. J. 2011, 17, 4968. doi:10.1002/chem.201100139
Return to citation in text: [1] -
Bélanger, D.; Tong, X.; Soumaré, S.; Dory, Y. L.; Zhao, Y. Chem.–Eur. J. 2009, 15, 4428. doi:10.1002/chem.200802337
Return to citation in text: [1] -
Spangenberg, T.; Schoenfelder, A.; Breit, B.; Mann, A. Eur. J. Org. Chem. 2010, 6005. doi:10.1002/ejoc.201000865
Return to citation in text: [1] -
Paul, A.; Bittermann, H.; Gmeiner, P. Tetrahedron 2006, 62, 8919. doi:10.1016/j.tet.2006.07.007
We used the Dess–Martin oxidation and the Ohira–Bestmann reagent instead of the reported Swern oxidation and Corey–Fuchs olefination.
Return to citation in text: [1] -
Crabbé, P.; Fillion, H.; André, D.; Luche, J.-L. J. Chem. Soc., Chem. Commun. 1979, 859. doi:10.1039/C39790000859
Return to citation in text: [1] -
Kuang, J.; Ma, S. J. Org. Chem. 2009, 74, 1763. doi:10.1021/jo802391x
Return to citation in text: [1] -
Decker, M. Curr. Med. Chem. 2011, 18, 1464. doi:10.2174/092986711795328355
Compounds 3d–f and 4d–f can be considered as hybrid scaffolds, i.e., as a combination of the biologically relevant β-lactam and oxazinone frameworks. See for a review on hybrid chemical entities.
Return to citation in text: [1] -
Tsogoeva, S. B. Mini-Rev. Med. Chem. 2010, 10, 773. doi:10.2174/138955710791608280
Return to citation in text: [1] -
Meunier, B. Acc. Chem. Res. 2008, 41, 69. doi:10.1021/ar7000843
Return to citation in text: [1] -
Tietze, L. F.; Bell, H. P.; Chandrasekhar, S. Angew. Chem., Int. Ed. 2003, 42, 3996. doi:10.1002/anie.200200553
Return to citation in text: [1] -
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi:10.1021/cr900211p
Fused tricyclic 3,4-dihydro-2H-1,3-oxazin-2-one 4j was more efficiently obtained by performing the reaction at 80 °C. Fused indole derivatives are represented in numerous natural alkaloids and synthetic pharmaceuticals, which display a number of interesting biological activities.
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608. doi:10.1002/anie.200901843
Return to citation in text: [1] -
Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875. doi:10.1021/cr0505270
Return to citation in text: [1] -
Except for fused 6-methylene-1,3-oxazinan-2-one, 3j, heterocycles 3 or 4 were exclusively obtained. Surprisingly, the exposure of tert-butyl allenic carbamate 2j to the gold-catalyzed conditions at room temperature afforded the 1,3-oxazin-2-one 4j as the major adduct, with the corresponding 1,3-oxazinan-2-one adduct 3j being the minor component.
Return to citation in text: [1] -
Chen, B.; Wang, N.; Fan, W.; Ma, S. Org. Biomol. Chem. 2012, 10, 8465. doi:10.1039/c2ob26291f
Return to citation in text: [1] -
Shu, W.; Yu, Q.; Jia, G.; Ma, S. Chem.–Eur. J. 2011, 17, 4720. doi:10.1002/chem.201003611
Return to citation in text: [1] -
Lee, P. H.; Kang, D.; Choi, S.; Kim, S. Org. Lett. 2011, 13, 3470. doi:10.1021/ol2012132
Return to citation in text: [1] -
Mo, J.; Kang, D.; Eom, D.; Kim, S. H.; Lee, P. H. Org. Lett. 2013, 15, 26. doi:10.1021/ol3029274
Return to citation in text: [1] -
Lee, P. H.; Kim, S.; Kang, D.; Park, A.; Chary, B. C.; Kim, S. Angew. Chem., Int. Ed. 2010, 49, 6806. doi:10.1002/anie.201001799
Return to citation in text: [1] -
Komiya, S.; Ozaki, S. Chem. Lett. 1988, 17, 1431. doi:10.1246/cl.1988.1431
See for the isolation of an allylic gold complex.
Return to citation in text: [1] -
1H NMR of the reaction mixtures revealed the presence of 2-methylprop-1-ene, which is likely formed from the tert-butyl fragmentation. For pioneering work on the introduction of tert-butyl carbonates as a cation-trapping group in gold catalysis, see references [76-78].
Return to citation in text: [1] -
Buzas, A.; Gagosz, F. Org. Lett. 2006, 8, 515. doi:10.1021/ol053100o
Return to citation in text: [1] [2] -
Kang, J.-E.; Shin, S. Synlett 2006, 717. doi:10.1055/s-2006-933110
Return to citation in text: [1] [2] -
Shin, S.-H. Bull. Korean Chem. Soc. 2005, 26, 1925. doi:10.5012/bkcs.2005.26.12.1925
Return to citation in text: [1] [2] -
The corresponding second-order perturbation energy from the NBO method associated with this LP(O) → d(Au) delocalization was computed to be ca. −5 kcal/mol.
Return to citation in text: [1]
| 75. | 1H NMR of the reaction mixtures revealed the presence of 2-methylprop-1-ene, which is likely formed from the tert-butyl fragmentation. For pioneering work on the introduction of tert-butyl carbonates as a cation-trapping group in gold catalysis, see references [76-78]. |
| 76. | Buzas, A.; Gagosz, F. Org. Lett. 2006, 8, 515. doi:10.1021/ol053100o |
| 77. | Kang, J.-E.; Shin, S. Synlett 2006, 717. doi:10.1055/s-2006-933110 |
| 78. | Shin, S.-H. Bull. Korean Chem. Soc. 2005, 26, 1925. doi:10.5012/bkcs.2005.26.12.1925 |
| 71. | Lee, P. H.; Kang, D.; Choi, S.; Kim, S. Org. Lett. 2011, 13, 3470. doi:10.1021/ol2012132 |
| 72. | Mo, J.; Kang, D.; Eom, D.; Kim, S. H.; Lee, P. H. Org. Lett. 2013, 15, 26. doi:10.1021/ol3029274 |
| 73. | Lee, P. H.; Kim, S.; Kang, D.; Park, A.; Chary, B. C.; Kim, S. Angew. Chem., Int. Ed. 2010, 49, 6806. doi:10.1002/anie.201001799 |
| 74. |
Komiya, S.; Ozaki, S. Chem. Lett. 1988, 17, 1431. doi:10.1246/cl.1988.1431
See for the isolation of an allylic gold complex. |
| 1. |
Robles-Machín, R.; Adrio, J.; Carretero, J. C. J. Org. Chem. 2006, 71, 5023. doi:10.1021/jo060520y
See for the synthesis of oxazinones from N-Boc-(3-butyn)-1-amines. |
| 15. | Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074. doi:10.1002/anie.201101460 |
| 16. | Rivera-Fuentes, P.; Diederich, F. Angew. Chem., Int. Ed. 2012, 51, 2818. doi:10.1002/anie.201108001 |
| 17. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994. doi:10.1021/cr1004088 |
| 18. | Alcaide, B.; Almendros, P. Chem. Rec. 2011, 11, 311. doi:10.1002/tcr.201100011 |
| 19. | Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Soc. Rev. 2010, 39, 783. doi:10.1039/b913749a |
| 20. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45. doi:10.1021/ar800011h |
| 21. | Krause, N., Ed. Compounds with all carbon functions: Cumulenes and Allenes; Science of Synthesis; Georg Thieme: Stuttgart, Germany, 2007; Vol. 44. |
| 22. | Ma, S. Chem. Rev. 2005, 105, 2829. doi:10.1021/cr020024j |
| 23. | Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004. |
| 24. | Alcaide, B.; Almendros, P. Eur. J. Org. Chem. 2004, 3377. doi:10.1002/ejoc.200400023 |
| 25. | Bates, R. W.; Satcharoen, V. Chem. Soc. Rev. 2002, 31, 12. doi:10.1039/b103904k |
| 26. | Zimmer, R.; Dinesh, C. U.; Nandanan, E.; Khan, F. A. Chem. Rev. 2000, 100, 3067. doi:10.1021/cr9902796 |
| 27. | Hashmi, A. S. K. Angew. Chem., Int. Ed. 2000, 39, 3590. doi:10.1002/1521-3773(20001016)39:20<3590::AID-ANIE3590>3.0.CO;2-L |
| 69. | Chen, B.; Wang, N.; Fan, W.; Ma, S. Org. Biomol. Chem. 2012, 10, 8465. doi:10.1039/c2ob26291f |
| 70. | Shu, W.; Yu, Q.; Jia, G.; Ma, S. Chem.–Eur. J. 2011, 17, 4720. doi:10.1002/chem.201003611 |
| 8. | Richter, H.; Fröhlich, R.; Daniliuc, C.-G.; García-Mancheño, O. Angew. Chem., Int. Ed. 2012, 51, 8656. doi:10.1002/anie.201202379 |
| 9. | Kuznetsov, N. Yu.; Maleev, V. I.; Khrustalev, V. N.; Mkrtchyan, A. F.; Godovikov, I. A.; Strelkova, T. V.; Bubnov, Y. N. Eur. J. Org. Chem. 2012, 334. doi:10.1002/ejoc.201101114 |
| 10. | Shpak-Kraievskyi, P.; Yin, B.; Martel, A.; Dhal, R.; Dujardin, G.; Laurent, M. Y. Tetrahedron 2012, 68, 2179. doi:10.1016/j.tet.2012.01.002 |
| 11. | Tian, C.; Jiao, X.; Liu, X.; Li, R.; Dong, L.; Liu, X.; Zhang, Z.; Xu, J.; Xu, M.; Xie, P. Tetrahedron Lett. 2012, 53, 4892. doi:10.1016/j.tetlet.2012.07.011 |
| 12. | Zhou, H.-B.; Lee, J. H.; Mayne, C. G.; Carlson, K. E.; Katzenellenbogen, J. A. J. Med. Chem. 2010, 53, 3349. doi:10.1021/jm100052k |
| 13. | Sato, S.; Shibuya, M.; Kanoh, N.; Iwabuchi, Y. Chem. Commun. 2009, 6264. doi:10.1039/b913770j |
| 14. | Osa, Y.; Hikima, Y.; Sato, Y.; Takino, K.; Ida, Y.; Hirono, S.; Nagase, H. J. Org. Chem. 2005, 70, 5737. doi:10.1021/jo0501644 |
| 44. | Hashmi, A. S. K.; Schuster, A. M.; Litters, S.; Rominger, F.; Pernpointner, M. Chem.–Eur. J. 2011, 17, 5661. doi:10.1002/chem.201100132 |
| 1. |
Robles-Machín, R.; Adrio, J.; Carretero, J. C. J. Org. Chem. 2006, 71, 5023. doi:10.1021/jo060520y
See for the synthesis of oxazinones from N-Boc-(3-butyn)-1-amines. |
| 59. | Crabbé, P.; Fillion, H.; André, D.; Luche, J.-L. J. Chem. Soc., Chem. Commun. 1979, 859. doi:10.1039/C39790000859 |
| 60. | Kuang, J.; Ma, S. J. Org. Chem. 2009, 74, 1763. doi:10.1021/jo802391x |
| 2. | Eichner, S.; Knobloch, T.; Floss, H. G.; Fohrer, J.; Harmrolfs, K.; Hermane, J.; Schulz, A.; Sasse, F.; Spiteller, P.; Taft, F.; Kirschning, A. Angew. Chem., Int. Ed. 2012, 51, 752. doi:10.1002/anie.201106249 |
| 3. | Wolfe, A. L.; Duncan, K. K.; Parelkar, N. K.; Weir, S. J.; Vielhauer, G. A.; Boger, D. L. J. Med. Chem. 2012, 55, 5878. doi:10.1021/jm300330b |
| 4. | Taft, F.; Harmrolfs, K.; Nickeleit, I.; Heutling, A.; Kiene, M.; Malek, N.; Sasse, F.; Kirschning, A. Chem.–Eur. J. 2012, 18, 880. doi:10.1002/chem.201101640 |
| 5. | Chinkov, N.; Warm, A.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 2957. doi:10.1002/anie.201006689 |
| 6. | Fishkin, N.; Maloney, E. K.; Chari, R. V. J.; Singh, R. Chem. Commun. 2011, 47, 10752. doi:10.1039/c1cc14164c |
| 7. | Zhang, F.; Qian, L.; Flood, P. M.; Shi, J.-S.; Hong, J.-S.; Gao, H.-M. J. Pharmacol. Exp. Ther. 2010, 333, 822. doi:10.1124/jpet.110.165829 |
| 61. |
Decker, M. Curr. Med. Chem. 2011, 18, 1464. doi:10.2174/092986711795328355
Compounds 3d–f and 4d–f can be considered as hybrid scaffolds, i.e., as a combination of the biologically relevant β-lactam and oxazinone frameworks. See for a review on hybrid chemical entities. |
| 62. | Tsogoeva, S. B. Mini-Rev. Med. Chem. 2010, 10, 773. doi:10.2174/138955710791608280 |
| 63. | Meunier, B. Acc. Chem. Res. 2008, 41, 69. doi:10.1021/ar7000843 |
| 64. | Tietze, L. F.; Bell, H. P.; Chandrasekhar, S. Angew. Chem., Int. Ed. 2003, 42, 3996. doi:10.1002/anie.200200553 |
| 65. |
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi:10.1021/cr900211p
Fused tricyclic 3,4-dihydro-2H-1,3-oxazin-2-one 4j was more efficiently obtained by performing the reaction at 80 °C. Fused indole derivatives are represented in numerous natural alkaloids and synthetic pharmaceuticals, which display a number of interesting biological activities. |
| 66. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608. doi:10.1002/anie.200901843 |
| 67. | Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875. doi:10.1021/cr0505270 |
| 68. | Except for fused 6-methylene-1,3-oxazinan-2-one, 3j, heterocycles 3 or 4 were exclusively obtained. Surprisingly, the exposure of tert-butyl allenic carbamate 2j to the gold-catalyzed conditions at room temperature afforded the 1,3-oxazin-2-one 4j as the major adduct, with the corresponding 1,3-oxazinan-2-one adduct 3j being the minor component. |
| 49. | Singh, S.; Elsegood, M. R. J.; Kimber, M. C. Synlett 2012, 565. doi:10.1055/s-0031-1290335 |
| 56. | Bélanger, D.; Tong, X.; Soumaré, S.; Dory, Y. L.; Zhao, Y. Chem.–Eur. J. 2009, 15, 4428. doi:10.1002/chem.200802337 |
| 57. | Spangenberg, T.; Schoenfelder, A.; Breit, B.; Mann, A. Eur. J. Org. Chem. 2010, 6005. doi:10.1002/ejoc.201000865 |
| 43. | Higginbotham, M. C. M.; Bebbington, M. W. P. Chem. Commun. 2012, 48, 7565. doi:10.1039/c2cc33711h |
| 44. | Hashmi, A. S. K.; Schuster, A. M.; Litters, S.; Rominger, F.; Pernpointner, M. Chem.–Eur. J. 2011, 17, 5661. doi:10.1002/chem.201100132 |
| 45. | LaLonde, R. L.; Wang, Z. J.; Mba, M.; Lackner, A. D.; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 598. doi:10.1002/anie.200905000 |
| 46. | Li, H.; Widenhoefer, R. A. Org. Lett. 2009, 11, 2671. doi:10.1021/ol900730w |
| 47. | Winter, C.; Krause, N. Angew. Chem., Int. Ed. 2009, 48, 6339. doi:10.1002/anie.200902355 |
| 48. | Yeom, H.-S.; Lee, E.-S.; Shin, S. Synlett 2007, 2292. doi:10.1055/s-2007-985571 |
| 58. |
Paul, A.; Bittermann, H.; Gmeiner, P. Tetrahedron 2006, 62, 8919. doi:10.1016/j.tet.2006.07.007
We used the Dess–Martin oxidation and the Ohira–Bestmann reagent instead of the reported Swern oxidation and Corey–Fuchs olefination. |
| 42. | Alcaide, B.; Almendros, P. Adv. Synth. Catal. 2011, 353, 2561. doi:10.1002/adsc.201100160 |
| 79. | The corresponding second-order perturbation energy from the NBO method associated with this LP(O) → d(Au) delocalization was computed to be ca. −5 kcal/mol. |
| 28. | Hashmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. |
| 29. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448. doi:10.1039/c1cs15279c |
| 30. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657. doi:10.1021/cr100414u |
| 31. | Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536. doi:10.1039/c1cc10780a |
| 32. | Alcaide, B.; Almendros, P.; Alonso, J. M. Org. Biomol. Chem. 2011, 9, 4405. doi:10.1039/c1ob05249g |
| 33. | Bandini, M. Chem. Soc. Rev. 2011, 40, 1358. doi:10.1039/c0cs00041h |
| 34. | Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078 |
| 35. | Lipshutz, B.; Yamamoto, Y., Eds. Coinage Metals in Organic Synthesis. Chem. Rev. 2008, 108, 2793–3442. |
| 36. | Hutchings, G. J.; Brust, M.; Schmidbaur, H., Eds. Gold – Chemistry, Materials and Catalysis. Chem. Soc. Rev. 2008, 37, 1745–2140. |
| 37. | Muzart, J. Tetrahedron 2008, 64, 5815. doi:10.1016/j.tet.2008.04.018 |
| 38. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x |
| 39. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333. doi:10.1039/b612008c |
| 40. | Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896. doi:10.1002/anie.200602454 |
| 41. |
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271. doi:10.1002/adsc.200600368
See for a review. |
| 50. | Alcaide, B.; Almendros, P.; Quirós, M. T.; López, R.; Menéndez, M. I.; Sochacka-Ćwikła, A. J. Am. Chem. Soc. 2013, 135, 898. doi:10.1021/ja3108966 |
| 51. | Alcaide, B.; Almendros, P.; Cembellín, S.; Martínez del Campo, T.; Fernández, I. Chem. Commun. 2013, 49, 1282. doi:10.1039/c2cc37872h |
| 52. | Alcaide, B.; Almendros, P.; Alonso, J. M.; Fernández, I. Chem. Commun. 2012, 48, 6604. doi:10.1039/c2cc32015k |
| 53. | Alcaide, B.; Almendros, P.; Aragoncillo, C.; Gómez-Campillos, G.; Arnó, M.; Domingo, L. R. ChemPlusChem 2012, 77, 563. doi:10.1002/cplu.201200090 |
| 54. | Alcaide, B.; Almendros, P.; Alonso, J. M.; Quirós, M. T.; Gadziński, P. Adv. Synth. Catal. 2011, 353, 1871. doi:10.1002/adsc.201100209 |
| 55. | Alcaide, B.; Almendros, P.; Carrascosa, R. Chem.–Eur. J. 2011, 17, 4968. doi:10.1002/chem.201100139 |
| 76. | Buzas, A.; Gagosz, F. Org. Lett. 2006, 8, 515. doi:10.1021/ol053100o |
| 77. | Kang, J.-E.; Shin, S. Synlett 2006, 717. doi:10.1055/s-2006-933110 |
| 78. | Shin, S.-H. Bull. Korean Chem. Soc. 2005, 26, 1925. doi:10.5012/bkcs.2005.26.12.1925 |
© 2013 Alcaide et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)