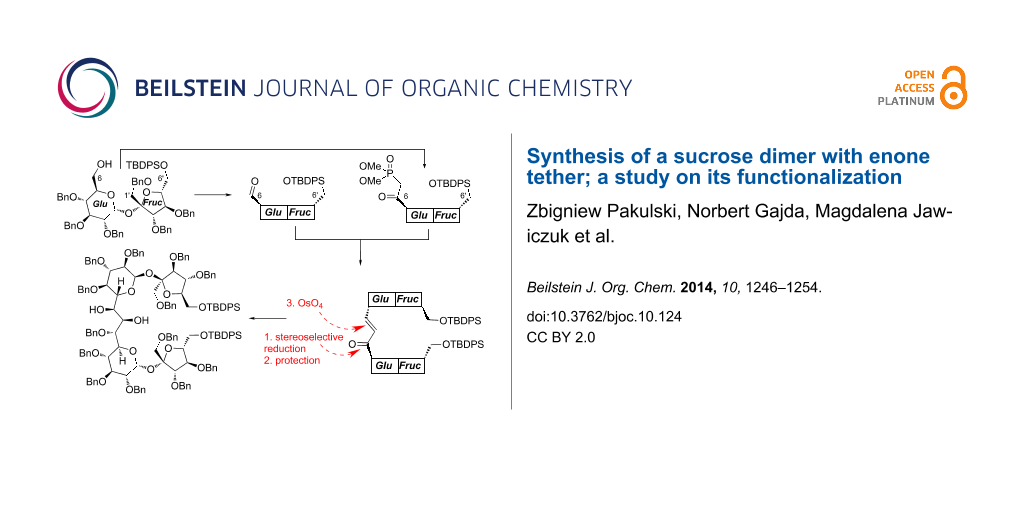
Abstract
The reaction of appropriately functionalized sucrose phosphonate with sucrose aldehyde afforded a dimer composed of two sucrose units connected via their C6-positions (‘the glucose ends’). The carbonyl group in this product (enone) was stereoselectively reduced with zinc borohydride and the double bond (after protection of the allylic alcohol formed after reduction) was oxidized with osmium tetroxide to a diol. Absolute configurations of the allylic alcohol as well as the diol were determined by circular dichroism (CD) spectroscopy using the in situ dimolybdenum methodology.

Graphical Abstract
Introduction
Molecular recognition is one of the most important phenomena in stereoselective processes. Chiral crown ethers (or analogs) are particularly useful in enantioselective reactions [1,2] as well as differentiation of chiral guests [3,4]. From all of the chiral platforms designed for such receptors, sugars are the most promising due to their availability and biocompatibility. Up to date only monosaccharides have found a wide application in the synthesis of crown ether analogs [5,6]. The disaccharide scaffold is much less pronounced [7].
During the past decade we have become engaged in the preparation of the analogs of crown and aza-crown ethers with sucrose scaffold. It is based on a selective protection of 1’,2,3,3’,4,4’-hexa-O-benzylsucrose (1) either at the glucose (C-6) [8] or fructose (C-6’) [9] end and further transformations to a variety of macrocycles (2–4; Figure 1). Such receptors exhibit interesting complexing properties towards chiral ammonium salts including amino acids [10-14]. More complex sucrose macrocycles, such as 5, are available, although in rather low yield [15].
![[1860-5397-10-124-1]](/bjoc/content/figures/1860-5397-10-124-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of sucrose-based macrocycles.
Figure 1: Examples of sucrose-based macrocycles.
In this paper we present an approach to other derivatives containing two sucrose units. This type of dimers may be eventually used for the construction of macrocycles by (simple) connecting their C-6’ (fructose) ends.
Results and Discussion
Coupling of two sugar units can be performed by a number of methods. The best one in our hands was the Wittig-type methodology shown in Figure 2. The properly activated sugar is converted into phosphorane or phosphonate which – upon reaction with an aldehyde derived from another monosaccharide – provides higher carbon sugar (HCS) enone [16-18].
![[1860-5397-10-124-2]](/bjoc/content/figures/1860-5397-10-124-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesis of higher sugar precursors by a Wittig-type methodology.
Figure 2: Synthesis of higher sugar precursors by a Wittig-type methodology.
Application of this methodology to selectively protected 2,3,3’,4,4’-penta-O-benzylsucrose allowed us to elongate the parent disaccharide at either terminal position (1’,6’, and 6’) by relatively small (C2 or C7) unit and prepare so-called higher sucroses in good yields [19,20]. A similar approach is used now for a more convenient hexa-O-benzyl derivative which is easily silylated at the ‘fructose end’ providing alcohol 6 [8]. This alcohol was converted into aldehyde 7 [21] (route a in Scheme 1) and separately into phosphonate 9 (route b). Reaction of both synthons under the mild PTC conditions [22-24] afforded the respective enone 10 in good yield (Scheme 1).
![[1860-5397-10-124-i1]](/bjoc/content/inline/1860-5397-10-124-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of higher sugar enone 10.
Scheme 1: Synthesis of higher sugar enone 10.
Functionalization of the three-carbon atom unit connecting the C5-positions of both sucrose units required reduction of the carbonyl group of the enone system and oxidation of the double bond.
We have already reported that reduction of higher carbon sugar enones of the D series with zinc borohydride is highly selective and provides the corresponding allylic alcohols with the R configuration at the newly created stereogenic center, as the only products. This can be rationalized assuming the cyclic model of such reduction [25] shown in Scheme 2. We expected, therefore, also very high selectivity in the reduction of 10.
![[1860-5397-10-124-i2]](/bjoc/content/inline/1860-5397-10-124-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the diol 13 containing two sucrose units.
Scheme 2: Synthesis of the diol 13 containing two sucrose units.
Indeed, treatment of enone 10 with Zn(BH4)2 under the standard conditions afforded allylic alcohol 11 as single stereoisomer in 65% yield. Based on our model, the R-configuration might be safely assigned to the new stereogenic center. This assignment was further verified independently by circular dichroism spectroscopy (CD) using the in situ dimolybdenum methodology (see next chapter).
Next steps of the synthesis consisted of the protection of the C6–OH as benzyl ether (to 12) and osmylation of the double bond. The cis-dihydroxylation provided, as single stereoisomer, a diol to which structure 13 could be assigned on the basis of the Kishi rule [26] (Scheme 2). It postulates that the attack of OsO4 occurs from the side opposite to hydroxy (alkoxy) substituent(s) flanking the double bond. Since in 12, both alkoxy units act in the same direction, very high diastereoselectivity is not surprising. The assignment of the configuration of this diol was further confirmed also by the CD methodology; this is discussed in the next chapter.
Determination of the absolute configuration of 11 and 13
It is widely acceptable that the circular dichroism (CD) spectroscopy utilizing the in situ dimolybdenum methodology offers the hard proof of the absolute configuration of the vic-diols [27-29]. In this methodology, dimolybdenum tetraacetate acts as auxiliary chromophore allowing the application of electronic circular dichroism (ECD) to (otherwise in ECD non-observable) vic-diols. Mo2(OAc)4 when mixed with a chiral diol ligand forms complexes active in ECD in which a transfer of ligand chirality to the in situ-formed complex occurs in solution. Thus, stereochemistry of vic-diols can be easily assigned based on the helicity rule developed for this class of compounds. This rule correlates the positive/negative signs of Cotton effects (CE) occurring in the 300–400 nm spectral range in the ECD spectra with the positive/negative sign of the O–C–C–O torsion angle of the diol unit of resultant complexes with the Mo2-core. The basic assumption leading to the assignment of the absolute configuration (AC) based only on the ECD spectra with the Mo2-core preferring the gauche conformation of the diol units with both O–C–C–C fragments in an antiperiplanar arrangement (Figure 3). This arrangement is favored, for steric reasons, i.e., to avoid any interaction with the carboxylate ligands remaining in the stock complex. As a result of the structure–ECD spectra relationship, it is possible to assign the AC of the diol moiety unambiguously on the basis of the ECD spectra alone.
![[1860-5397-10-124-3]](/bjoc/content/figures/1860-5397-10-124-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CD spectra of in situ formed chiral complexes of 13 (green line), 14 (purple line) and 16 (blue line) with dimolybdenum tetraacetate recorded in DMSO; right: preferred gauche conformation of the diols 13, 14 and 16 in the complex with Mo2-core.
Figure 3: CD spectra of in situ formed chiral complexes of 13 (green line), 14 (purple line) and 16 (blue lin...
In the past few years, this simple but, above all, efficient and effective method is becoming more and more recognized as evidenced by the steadily increasing number of reports in the literature about its successful application in the determination of the AC of 1,2-diols [30-32].
Therefore, in assignment of the AC of compounds under the present study (13, 14 and 16), we decided just to take advantage of the in situ methodology.
This method was used to prove indirectly the 6R configuration at the newly created stereogenic center in allylic alcohol 11. The double bond in 11 was cleaved with ozone and the resulting ozonide was reduced with NaBH4; this sequence afforded sucrose vic-diol 14 and (as a byproduct) sucrose alcohol 6 (Scheme 3).
![[1860-5397-10-124-i3]](/bjoc/content/inline/1860-5397-10-124-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of model sucrose diols.
Scheme 3: Synthesis of model sucrose diols.
The positive sign of the Cotton effect at around 307.0 nm recorded for the complex of the diol 14 with Mo2(OAc)4 unambiguously pointed at the 6R configuration (Figure 3). However, to exclude any errors we have also prepared 14 and epimeric alcohol 16; its synthesis is shown in Scheme 3.
First, aldehyde 7 was converted into olefin 15 by treatment with the simplest Wittig reagent: Ph3P=CH2. Subsequent osmylation of the double bond in 15 provided two stereoisomeric diols in a 1:1 ratio; the first one was identical in all respects with the diol obtained from degradation of 11.
The resultant ECD spectra of the Mo2-core with compounds 13 and 16 are shown in Figure 3. Based on the positive CE’s at 308.5 nm for 13 and negative at 310 nm for 16, respectively, the positive (negative) sign of the O–C–C–O torsion angle has been attributed to these diols. In the next step, based on the preferred gauche conformation of the diol unit with both O–C–C–C fragments in an antiperiplanar arrangement as shown in Figure 3, we were able to assign unambiguously the (7R,8R) AC to diol 13 and (6S) to 16.
Conclusion
Coupling of two properly activated sucrose sub-units afforded the dimer in which both glucose-rings were connected via an enone linker. The dimer was then converted into a (partially protected) triol via a stereoselective reduction of the carbonyl group and highly selective cis-dihydroxylation of the double bond. The configuration at each new stereogenic center was determined by CD spectroscopy using the so-called dimolybdenum methodology which allows for fast, easy, and effective assignment of the absolute configuration of vic-diols. We have confirmed the usefulness of this simple methodology which can be applied even in cases when other spectroscopic methods fail.
It is worthy to point out that this methodology, which is used in the synthesis of more simple derivatives such as higher carbon sugars, was also applicable for the preparation of the sucrose dimer.
Experimental
General methods
All reported NMR spectra were recorded with a Varian-Vnmrs-600 MHz spectrometer (at 600 and 150 MHz for 1H and 13C NMR spectra, respectively) for solutions in CDCl3 at room temperature. Chemical shifts (δ, ppm) were determined relative to TMS as the internal standard. Most of the resonances were assigned by COSY (1H–1H) and gradient selected HSQC and HMBC correlations. Mass spectra were recorded with an ESI/MS Mariner (PerSeptive Biosystem) mass spectrometer. Elemental analyses were obtained using a Perkin-Elmer 2400 CHN analyzer. Optical rotations were measured with a Jasco P-2000 digital polarimeter for solutions in CHCl3 (c = 0.3) at room temperature. Flash and column chromatographic separations were performed on silica gel (Merck, 230–400 mesh). Progress of the reactions was monitored by thin-layer chromatography (TLC) performed on aluminum plates covered with silica gel (60 F254, Merck).
The ECD spectra were acquired at room temperature in DMSO (for UV-spectroscopy, Fluka) on a Jasco J-715 spectropolarimeter and were collected at 0.5 nm/step with an integration time of 0.25 s over the range 235–800 nm with 200 nm/min scan speed, 5 scans. For the ECD standard measurements the chiral diols (~3.6 mg, ca. 0.003 M) was mixed with stock complex [Mo2(O2CCH3)4] (Mo1) (~0.9 mg, ca. 0.002 M) and dissolved in DMSO (1 mL) so that the molar ratio of the stock complex to ligand was about 1:1.5 in general. Quantitative values could not be obtained with the in situ dimolybdenum method since the concentration of the chiral complex formed in solution and its actual structure were unknown. So the ECD data are given as the Δε' values, which are calculated in the usual manner by means of the equation Δε' = ΔA/c × d, A being the absorption, c the molar concentration of the chiral ligand, assuming 100% complexation, and d the path length of the cell.
The numbering of the atoms in sucrose dimers is as followed. The bottom part (green) is marked A and the top marked as B. The original numbering of the sucrose skeleton in both parts is retained (i.e., the glucose part is numbered C1–C6 and fructose C1'–C6').
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-124-i4.svg?max-width=637&scale=1.18182)
Synthesis of phosphonate 9. To a cooled −78 °C solution of dimethyl methylphosphonate (110 μL, 1.0 mmol) in THF (7 mL) a 2.5 M solution of butyllithium in hexane (0.4 mL, 1.0 mmol) was added and the mixture was stirred for 15 min. Then, a solution of 8 (325 mg, 0.28 mmol) in THF (5 mL) was slowly added and the mixture was stirred for additional 30 min. Reaction was quenched by addition of a saturated solution of NaCl (5 drops). The mixture was concentrated, and the residue was purified by column chromatography (hexane–ethyl acetate, 10:1 → 1:1) to afford title compound 9 (227 mg, 65%) as a foam. [α]D20 16.2.; 1H NMR δ 7.64–7.61 (m, 4H, Ar), 7.35–7.18 (m, 36H, aryl-H), 6.08 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.86–4.38 (m, 12H, PhCH2), 4.54 (d, 1H, J5,4 = 9.9 Hz, H-5), 4.48 (d, 1H, J3,4 = 7.5 Hz, H-3'), 4.45 (dd, 1H, J4,3 = 7.5, J4,5 = 15.0 Hz, H-4'), 4.03 (dd, 1H, J6,5 = 3.8, J6,6’ 11.5 Hz, H-6'), 3.94 (m, 2H, H-3, H-5'), 3.85 (dd, 1H, J6,5 = 4.1 Hz, J6,6’ = 11.5 Hz, H-6'), 3.71 (dd, 1H, J4,3 = 9.1 Hz, J4,5 = 9.9 Hz, H-4), 3.67 (d, 1H, Jgem = 10.8 Hz, H-1'), 3.62 (d, 3H, JH,P = 11.1 Hz, OCH3), 3.61 (d, 3H, JH,P = 11.2 Hz, OCH3), 3.56 (d, 1H, Jgem = 10.8 Hz, H-1'), 3.43 (dd, 1H, J2,1 = 3.6, J2,3 = 9.7 Hz, H-2), 3.26 (dd, 1H, J7,7' = 15.7 Hz, JH,P = 20.3 Hz, H-7), 2.97 (dd, 1H, J7,7' = 15.7 Hz, JH,P = 20.8 Hz, H-7), 1.06 (s, 9H, t-Bu); 13C NMR δ 198.5 (d, J 7.4 Hz, C=O), 138.7, 138.5, 138.2, 138.1, 137.8, 137.7, 135.6, 135.4, 133.1, 132.7, 129.8, 129.8, 128.6–127.5 (Ar), 104.5 (C-2'), 89.2 (C-1), 83.3 (C-3'), 81.5 (C-3), 80.8 (C-4'), 80.5 (C-5'), 79.5 (C-2), 77.9 (C-4), 75.7 (PhCH2), 74.9 (d, J 5.0 Hz, C-5), 74.8 (PhCH2), 73.5 (PhCH2), 73.4 (PhCH2), 72.9 (PhCH2), 72.0 (PhCH2), 72.0 (C-1'), 63.5 (C-6'), 52.8 (d, J = 6.3 Hz, OCH3), 52.6 (d, J = 6.4 Hz, OCH3), 37.9 (d, J = 136.9 Hz, C-7), 26.9 (t-Bu-CH3), 19.2 (t-Bu-C); 31P NMR (CDCl3) δ 23.5; anal. calcd for C73H81O14PSi (1241.51): C, 70.62; H, 6.58; found: C, 70.44; H, 6.79.
Synthesis of ketone 10. To a solution of aldehyde 7 (200 mg, 0.18 mmol), phosphonate 9 (220 mg, 0.18 mmol), and 18-crown-6 (70 mg) in toluene (25 mL) potassium carbonate (300 mg) was added and the suspension was stirred at rt for 4 days. The solvents were evaporated and the residue was purified by column chromatography (hexane–ethyl acetate, 40:1 → 5:1) to afford title compound 10 as a foam (287 mg; 73%). [α]D20 41.8; 1H NMR δ 7.62–7.61 (m, 8H, Ar), 7.28–7.06 (m, 73H, Ar, =CH-CO), 6.75 (dd, 1H, J = 1.8 Hz, 15.7 Hz, =CH), 5.90 (d, 1H, J = 3.5 Hz, H-1A), 5.82 (d, 1H, J = 3.5 Hz, H-1B), 4.74–4.26 (m, 24H, 12 × PhCH2), 4.67 (d, 1H, J = 10.2 Hz, H-5A), 4.58 (d, 1H, J = 12.3 Hz, H-5B), 4.37–4.26 (m, 4H, furanose), 4.00–3.80 (m, 8H, H-3A, H-3B, 4 × H-6', 2 × furanose), 3.77 (d, 1H, J = 11.0 Hz, H-1'B), 3.68 (d, 1H, J = 10.9 Hz, H-1'A), 3.50 (dd, 1H, J = 9.1 Hz, 10.0 Hz, H-4A), 3.46 (d, 1H, J = 10.9 Hz, H-1'A), 3.42–3.38 (m, 2H, H-1'B, H-2A), 3.25 (dd, 1H, J = 3.5 Hz, 9.6 Hz, H-2A), 2.97 (dd, 1H, J = 9.1 Hz, 9.9 Hz, H-4B), 1.03 (s, 9H, t-Bu), 1.02 (s, 9H, t-Bu); 13C NMR δ 195.2 (C=O), 144.7 (=CH-CO), 138.9, 138.8, 138.4, 138.3, 138.2, 138.1, 138.1, 138.0, 137.9, 137.9, 137.8, 137.7, 135.6, 135.6, 135.5, 133.4, 133.3, 133.2, 133.1, 129.7, 129.7, 129.7, 129.6, 128.4–127.4 (Ar), 124.8 (=CH) 105.1 (C-2'A), 104.8 (C-2'B), 90.3 (C-1A), 90.2 (C-1B), 84.4, 83.9, 83.3, 83.2, 81.8 (C-4B), 81.7, 81.7, 81.5 (C-3A), 81.4 (C-3B), 79.8 (C-2A, C-2B), 79.6 (C-4A), 75.7 (PhCH2), 75.4 (PhCH2), 74.9 (C-5A, PhCH2), 74.4 (PhCH2), 73.5 (PhCH2), 73.4 (PhCH2), 73.1 (PhCH2), 73.0 (PhCH2), 72.6 (PhCH2), 72.4 (PhCH2), 72.1 (PhCH2),71.8 (PhCH2), 70.6 (C-1'), 70.4 (C-1'), 69.8 (C-5B), 65.3 (C-6'), 65.0 (C-6'), 27.0 (t-Bu), 27.0 (t-Bu), 19.3 (CH3), 19.3 (CH3); anal. calcd for C141H148O21Si2 (2234.91): C, 75.78; H, 6.68; found: C, 75.59; H, 6.80.
Stereoselective reduction of ketone 10. To an ice-cooled solution of ketone 10 (270 mg, 0.12 mmol) in Et2O (15 mL), an etheral solution of zinc borohydride (0.6 mmol) was added and the mixture was stirred at 0 °C for 1 h. Water (10 drops) was added to decompose excess of hydride, the solvents were evaporated, and the residue was purified by column chromatography (hexane–ethyl acetate, 10:1 → 5:1) to afford title compound 11 (176 mg, 65%) as a foam. [α]D20 40.4; 1H NMR δ 7.63–7.61 (m, 8H, Ar), 7.30–7.05 (m, 72H, Ar), 5.97 (dd, 1H, J = 7.9 Hz, 15.6 Hz, =CH-CHOH), 5.83 (d, 2H, J = 3.6 Hz, H-1A, H-1B), 5.76 (dd, 1H, J = 5.5 Hz, 15.6 Hz, =CH), 4.80–4.73 (m, 5H, PhCH2), 4.63–4.41 (m, 17H, PhCH2, H-3', H-5B), 4.39–4.34 (m, 6H, PhCH2, H-3', H-3', H-4', H-6A), 4.32–4.28 (m, 1H, H-4'), 4.25 (dd, 1H, J = 2.2 Hz, 10.3 Hz, H-5A), 4.13 (d, 1H, J = 10.9 Hz, PhCH2), 4.06–4.04 (m, 1H, H-5'), 3.99–3.95 (m, 2H, 2 × H-6'), 3.92–3.89 (m, 2H, H-5', H-6'), 3.86–3.79 (m, 3H, H-3A, H-1', H-3B), 3.65 (d, 1H, J = 11.0 Hz, H-1'), 3.52 (d, 1H, J = 11.0 Hz, H-1'), 3.49 (d, 1H, H-1'), 3.35 (dd, 1H, J = 3.5 Hz, 9.6 Hz, H-2B), 3.27–3.24 (m, 1H, H-4A), 3.21–3.16 (m, 2H, H-2A, H-4B), 1.03 (s, 9H, t-Bu), 1.02 (s, 9H, t-Bu); 13C NMR δ 139.0, 138.9, 138.6, 138.6, 138.5, 138.4, 138.3, 138.1, 138.0, 137.9, 137.8, 137.7, 135.7, 135.6, 135.5, 135.5, 133.5, 133.3, 133.3, 132.8, 131.3 (=CH), 130.4 (=CH-CHOH), 129.8, 129.7, 129.7, 129.6, 128.5–127.3 (Ar), 105.0 (C-2'), 104.5 (C-2'), 90.2 (C-1B), 89.0 (C-1A), 84.5 (C-3'), 83.6 (C-4'), 83.1 (C-4'), 82.3 (C-3A), 82.2 (C-4B), 81.8 (C-5'), 81.6 (C-3'), 81.5 (C-3B), 81.0 (C-5'), 80.4 (C-2A), 80.0 (C-2B), 78.4 (C-4A), 75.5 (PhCH2), 75.4 (PhCH2), 74.6 (PhCH2), 74.4 (PhCH2), 73.6 (PhCH2), 73.4 (PhCH2), 73.4 (C-5A), 73.1 (PhCH2), 73.0 (PhCH2), 72.7 (PhCH2), 72.6 (PhCH2), 71.8 (PhCH2), 71.7 (PhCH2), 71.5 (C-6A), 71.4 (C-1'), 70.5 (C-1'), 70.0 (C-5B), 65.8 (C-6'), 63.7 (C-6'), 27.0 (t-Bu), 27.0 (t-Bu), 19.3 (CH3), 19.3 (CH3); anal. calcd for C141H150O21Si2 (2236.92): C, 75.71; H, 6.76; found: C, 75.51; H, 6.63.
Benzylation of 11. To a solution of alcohol 11 (82 mg, 0.037 mmol) in DMF (1 mL), sodium hydride (60% suspension in mineral oil, 7 mg) was added and the mixture was stirred for 30 min at rt. Benzyl bromide (11 μL, 0.092 mmol) was added, and stirring was continued overnight. Excess of sodium hydride was decomposed with methanol (0.5 mL). The product was isolated by column chromatography (hexane–ethyl acetate, 10:1 → 5:1) to afford 12 (34 mg, 40%) as a foam. [α]D20 21.8 (c 0.9, CH2Cl2); 1H NMR δ 7.67–7.63 (m, 8H, Ar), 7.28–7.00 (m, 77H, Ar), 5.93–5.89 (m, 2H, H-1B, =CH-CHOH), 5.63–5.59 (m, 2H, H-1A, =CH), 4.79–4.50 (m, 13H, H-5B, PhCH2), 4.47–4.16 (m, 18H, H-5A, PhCH2), 4.10–3.75 (m, 12H, H-6A, 4 × H-6', H-3B, 2 × H-1', H-3A, 2 × furanose, PhCH), 3.56–3.52 (m, 2H, 2 × H-1'), 3.41 (dd, 1H, J = 3.6, 9.7 Hz, H-2B), 3.19–3.14 (m, 2H, H-4A, H-4B), 2.92 (dd, 1H, J = 3.5, 9.6 Hz, H-2A), 1.03 (s, 9H, t-Bu), 1.03 (s, 9H, t-Bu); 13C NMR δ: 139.0, 138.8, 138.7, 138.6, 138.5, 138.4, 138.3, 138.3, 138.3, 138.3, 138.2, 138.0, 137.8, 135.6, 135.5, 135.5, 135.5, 134.4 (=CH), 133.7, 133.6, 133.4, 133.2, 129.7, 129.6, 129.6, 129.6, 128.3–127.0 (Ar), 104.4 (C-2'), 104.4 (C-2'), 90.1 (C-1A), 89.9 (C-1B), 84.5, 84.1, 83.2, 82.2, 82.0 (C-3A), 82.0 (C-4B), 81.6 (C-3B, C-6A), 80.4 (C-2A), 79.9 (C-2B), 78.6, 78.2 (C-4A), 75.6 (PhCH2), 75.2 (PhCH2), 74.6 (PhCH2), 74.2 (PhCH2), 73.6 (PhCH2), 73.2 (PhCH2), 73.1 (PhCH2), 73.0 (PhCH2), 72.7 (PhCH2), 72.6 (C-5A), 72.5 (PhCH2), 71.9 (PhCH2), 71.8 (PhCH2), 71.0 (C-1'), 70.3, 70.2 (C-1'), 69.9, 66.7 (C-6'), 65.6 (C-6'), 27.0 (t-Bu), 26.9 (t-Bu), 19.3 (CH3), 19.3 (CH3); anal. calcd for C148H156O21Si2 (2327.05): C, 76.39; H, 6.76; found: C, 76.44; H, 6.73.
Dihydroxylation of the double bond of 12. Olefin 12 (60 mg, 0.026 mmol) and OsO4 (30 mg, 0.120 mmol) were dissolved in pyridine (4 mL), and stirred for 48 h. The solvent was evaporated, the residue was dissolved in ethyl acetate (10 mL), to which sat. aq Na2S2O3 (1 mL) was added, and the suspension was stirred for 2 days. The organic layer was separated, solvents were evaporated to dryness and the residue was purified by column chromatography (hexane–ethyl acetate, 5:1) to afford 13 (44 mg, 73%) as colorless glass. [α]D20 22.8; 1H NMR δ 7.64–7.59 (m, 8H, Ar), 7.27–6.98 (m, 77H, Ar), 5.83 (bs, 1H, H-1A), 5.80 (d, 1H, J1,2 = 3.5 Hz, H-1B), 4.82–4.69 (m, 5H, PhCH2), 4.65–4.49 (m, 11H, PhCH2, sugar-H), 4.45–4.34 (m, 10H, PhCH2, 2 × sugar-H), 4.29–4.23 (m, 7H, PhCH2, 5 × sugar-H), 4.08–4.05 (m, 2H, PhCH2, sugar-H), 4.01–3.95 (m, 6H, 3 × H-6', 3 × sugar-H), 3.89–3.78 (m, 4H, H-1', H-3A, H-3B, H-6'), 3.67 (d, 1H, J = 11.1 Hz, H-1'), 3.58–3.55 (m, 2H, H-1', H-1'), 3.37 (dd, 1H, J = 3.6 Hz, 9.6 Hz, H-2B), 3.36–3.32 (m, 2H, H-2A, sugar-H), 1.02 (s, 9H, t-Bu), 1.01 (s, 9H, t-Bu); 13C NMR δ 139.1, 139.0, 138.9, 138.8, 138.6, 138.3, 138.2, 138.2, 138.0, 137.9, 137.7, 137.6, 135.6, 135.6, 135.5, 135.5, 133.6, 133.4, 133.3, 132.9, 129.7, 129.7, 129.6, 129.5, 128.4-127.1 (Ar), 105.1 (C-2'), 104.7 (C-2'), 90.7 (C-1B), 89.4 (C-1A), 84.4, 83.5, 82.5 (C-3B), 82.0, 81.9, 81.6 (C-3A), 81.2, 80.4 (C-2B), 79.7 (C-2A), 79.3, 78.6, 75.5 (PhCH2), 75.3 (PhCH2), 74.6 (PhCH2), 74.3, 74.3 (PhCH2), 73.5 (PhCH2), 73.2 (PhCH2), 73.1 (PhCH2), 73.1 (PhCH2), 72.7 (PhCH2), 72.5 (PhCH2), 71.9 (PhCH2), 71.8 (PhCH2), 70.9 (C-1'), 70.7, 70.1 (C-1'), 69.7, 68.2, 66.4 (C-6'), 64.2 (C-6'), 27.0 (t-Bu), 19.3 (CH3), 19.2 (CH3); MS (ESI): 2383.08 [M + Na]+; anal. calcd for C148H158O23Si2 (2361.06): C, 75.29; H, 6.75; found: C, 75.29; H, 6.79.
Synthesis of olefin 15. To a suspension of methyltriphenylphosphonium bromide (895 mg, 2.50 mmol) in benzene (20 mL) a 2.5 M solution of BuLi in hexane (0.95 mL, 2.30 mmol) was added and the mixture was stirred at rt for 90 min. A solution of aldehyde 7 (298 mg, 0.27 mmol) in benzene (5 mL) was added, the mixture was stirred for another 45 min, quenched with water (100 μL), concentrated, and the residue was purified by column chromatography (hexane–ethyl acetate, 15:1 → 10:1) to afford 15 (172 mg, 58%) as a foam. [α]D20 24.0; 1H NMR δ 7.67–7.64 (m, 4H, Ar), 7.35–7.18 (m, 36H, Ar), 5.77–5.72 (m, 2H, H-1,6), 5.12–5.09 (m, 1H, H-7), 5.02–5.00 (m, 1H, H-7), 4.82–4.50 (m, 11H, PhCH2), 4.45–4.43 (m, 3H, H-3',5,PhCH), 4.30 (t, 1H, J4,3 = J4,5 = 7.4 Hz, H-4'), 4.05–4.02 (m, 1H, H-5'), 3.94 (dd, 1H, J6,5 = 4.8 Hz, J6,6’ = 11.1 Hz, H-6'), 3.90–3.87 (m, 2H, H-3,6'), 3.69 (d, 1H, Jgem = 11.0 Hz, H-1'), 3.50 (d, 1H, Jgem = 11.0 Hz, H-1'), 3.44 (dd, 1H, J2,1 = 3.7 Hz, J2,3 = 9.7 Hz, H-2), 3.16 (dd, 1H, J4,3 = 9.4 Hz, J4,5 = 9.5 Hz, H-4), 1.06 (s, 9H, t-Bu); 13C NMR δ 138.9, 138.6, 138.4, 138.3, 138.0, 135.7, 135.6, 135.6, 133.5, 133.3, 129.6, 129.6, 128.3–127.5 (Ar), 117.0 (C-7), 104.3 (C-2'), 89.2 (C-1), 83.8 (C-3'), 82.5 (C-4), 82.3 (C-4'), 81.6 (C-3), 81.2 (C-5'), 80.0 (C-2), 75.6 (PhCH2), 74.8 (PhCH2), 73.4 (PhCH2), 72.9 (PhCH2), 72.6 (PhCH2), 72.3 (PhCH2), 71.4 (C-1',5), 64.9 (C-6'), 26.9 (t-Bu-CH3), 19.3 (t-Bu-C); anal. calcd for C71H76O10Si (1117.47): C, 76.31; H, 6.86; found: C, 76.42; H, 6.99.
Ozonolytic cleavage of the double bond in sucrose dimer 11. Determination of the configuration at the carbinol center. Ozone was passed through a cooled solution of 11 (51 mg, 0.023 mmol) in CH2Cl2 (10 mL) until the blue color persisted (10 min). Dimethyl disulfide (210 μL) was added, the mixture was stirred for 10 min, concentrated, and the residue was dissolved in methanol (10 mL). Sodium borohydride (40 mg) was added, the mixture was stirred for 1 h, concentrated, and the crude product was purified by column chromatography (hexane–ethyl acetate, 7:3) to afford sucrose 6 (16 mg, 63%) and diol 14 (18 mg, 69%), both as foam.
Synthesis of diols 14 and 16. To a solution of 15 (145 mg, 0.13 mmol) in THF (10 mL), tert-butyl alcohol (500 μL), water (50 μL), NMO (80 mg, 0.68 mmol), and OsO4 (8 wt % in t-BuOH, 150 μL, 0.035 mmol) were added, and the mixture was stirred at rt for 18 h. Saturated aq Na2S2O3 (0.2 mL) was added, the mixture was stirred for 1 h at rt, concentrated, and the products were isolated by column chromatography (hexane–ethyl acetate, 5:1 → 3:1) to afford of 14 (60 mg; 40%) and 16 (58 mg 39%), both as foam.
Data for 14: [α]D20 32.6; 1H NMR δ 7.64–7.62 (m, 4H, aryl-H), 7.36–7.14 (m, 36H, Ar), 6.04 (d, 1H, J1,2 = 3.8 Hz, H-1), 4.95–4.39 (m, 10H, PhCH2), 4.47–4.45 (m, 4H, H-3',4',PhCH2), 4.14 (dd, 1H, J5,4 = 10.2, J5,6 = 4.1 Hz, H-5), 4.03 (dd, 1H, J6,5 = 3.4, J6,6’ = 11.6 Hz, H-6'), 3.93–3.89 (m, 2H, H-3,5'), 3.84–3.79 (m, 2H, H-6,6'), 3.65–3.62 (m, 2H, H-1',7), 3.55 (d, 1H, Jgem = 10.8 Hz, H-1'), 3.49–3.43 (m, 3H, H-2,4,7), 1.07 (s, 9H, t-Bu); 13C NMR δ 138.6, 138.2, 138.0, 137.9, 137.8, 137.3, 135.7, 135.4, 133.0, 132.6, 129.8, 129.7, 128.7–127.6 (Ar), 104.3 (C-2'), 88.2 (C-1), 82.9 (C-3'), 82.0 (C-3), 80.3 (C-5'), 80.1 (C-2,4'), 79.0 (C-4), 75.5 (PhCH2), 74.7 (PhCH2),73.4 (PhCH2), 73.1 (PhCH2), 72.8 (PhCH2), 72.3 (C-1'), 72.2 (C-6), 71.9 (PhCH2), 71.3 (C-5), 63.0 (C-6'), 62.9 (C-7), 26.9 (t-Bu-CH3), 19.2 (t-Bu-C); anal. calcd for C71H78O12Si (1151.49): C, 74.06; H, 6.83; found: C, 74.00; H, 6.79.
Data for 16: [α]D20 21.6; 1H NMR δ 7.64–7.62 (m, 4H, Ar), 7.37–7.16 (m, 36H, aryl-H), 6.04 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.93–4.40 (m, 12H, PhCH2), 4.40 (d, 1H, J = 6.5 Hz, H-3'), 4.32 (t, 1H, J4,3 = J4,5 = 6.7 Hz, H-4'), 4.03–3.99 (m, 2H, H-5,6'), 3.95 (t, 1H, J3,2 = J3,4 = 9.4 Hz, H-3), 3.92–3.91 (m, 1H, H-5'), 3.81 (dd, 1H, J6,5 = 4.0 Hz, J6,6’ = 11.4 Hz, H-6'), 3.74–3.72 (m, 1H, H-6), 3.70–3.66 (m, 2H, H-1',4), 3.55 (d, 1H, Jgem = 10.9 Hz, H-1'), 3.48–3.42 (m, 3H, H-2,7,7), 1.07 (s, 9H, t-Bu); 13C NMR δ 138.8, 138.5, 138.3, 137.9, 137.7, 137.5, 135.7, 135.5, 132.9, 132.7, 129.8, 129.8, 128.4–127.5 (Ar), 104.8 (C-2'), 89.3 (C-1), 83.2 (C-3'), 81.4 (C-3), 81.1 (C-4'), 80.8 (C-5'), 79.9 (C-2), 77.3 (C-4), 75.5 (PhCH2), 75.1 (PhCH2), 73.5 (PhCH2), 73.2 (PhCH2), 72.5 (PhCH2), 72.0 (PhCH2), 71.9 (C-5), 71.6 (C-1'), 68.9 (C-6), 64.5 (C-7), 63.1 (C-6'), 26.9 (t-Bu-CH3), 19.2 (t-Bu-C); anal. calcd for C71H78O12Si (1151.49): C, 74.06; H, 6.83; found: C, 73.94; H, 6.66.
Both compounds were further characterized as diacetates: 14-Ac and 16-Ac.
Data for 14-Ac: [α]D20 33.3; 1H NMR δ 7.66–7.64 (m, 4H, Ar), 7.34–7.21 (m, 36H, aryl-H), 5.82 (d, 1H, J1,2 = 3.6 Hz, H-1), 5.48 (m, 1H, H-6), 4.93–4.55 (m, 10H, PhCH2), 4.46–4.42 (m, 3H, H-3', PhCH2), 4.15 (dd, 1H, J5,4 = 10.3 Hz, J5,6 = 1.4 Hz, H-5), 4.03–3.96 (m, 3H, H-5', H-6', H-7), 3.91 (dd, 1H, J6,5 = 4.7, J6,6’ = 10.9 Hz, H-6'), 3.88 (t, 1H, J3,2 = J3,4 = 9.2 Hz, H-3), 3.67 (d, 1H, J1,1 = 11.0 Hz, H-1'), 3.51 (d, 1H, J1,1 = 11.0 Hz, H-1'), 3.49 (dd, 1H, J4,3 = 9.2 Hz, J4,5 = 10.3 Hz, H-4), 3.39 (dd, 1H, J2,1 = 3.6 Hz, J2,3 = 9.7 Hz, H-2), 1.96 (s, 3H, CH3), 1.84 (s, 3H, CH3), 1.05 (s, 9H, t-Bu); 13C NMR δ 170.5 (C=O), 169.8 (C=O), 138.6, 138.4, 138.2, 138.1, 138.0, 137.9, 135.7, 135.5, 133.3, 133.0, 129.7, 129.6, 128.3-127.5 (Ar), 104.2 (C-2'), 89.0 (C-1), 83.7 (C-3'), 82.0 (C-3), 81.9 (C-4'), 81.0 (C-5'), 79.8 (C-2), 77.6 (C-4), 75.6 (PhCH2), 74.6 (PhCH2), 73.3 (PhCH2), 73.1 (PhCH2), 72.8 (PhCH2), 72.1 (PhCH2), 71.4 (C-1'), 71.1 (C-6), 70.9 (C-5), 64.6 (C-6'), 63.2 (C-7), 26.9, 20.9, 20.8, 19.2; anal. calcd for C75H82O14Si × ½H2O (1253.57): C, 72.38; H, 6.72; found: C, 72.51; H, 6.36. HRMS (ESI) calc. for C75H86NO14Si [M + NH4]+: 1252.5818; found: 1252.5825.
Data for 16-Ac: [α]D20 13.1; 1H NMR (CDCl3) δ 7.65–7.63 (m, 4H, Ar), 7.34–7.20 (m, 36H, Ar), 6.09 (d, 1H, J1,2 = 3.5 Hz, H-1), 5.48 (ddd, 1H, J = 1.5 Hz, 2.9 Hz, 9.2 Hz, H-6), 4.88–4.42 (m, 12H, PhCH2), 4.42–4.38 (m, 2H, H-3', H-4'), 4.28 (dd, 1H, J7,6 = 9.1 Hz, J7,7 = 12.0 Hz, H-7), 4.11 (dd, 1H, J5,4 = 10.1 Hz, J5,6 = 1.3 Hz, H-5), 4.06 (dd, 1H, J7,6 = 3.1 Hz, J7,7 = 12.0 Hz, H-7), 4.02 (dd, 1H, J6,5 = 4.2 Hz, J6,6’ = 11.4 Hz, H-6'), 3.97–3.93 (m, 2H, H-3, H-5'), 3.85 (dd, 1H, J6,5 = 4.1 Hz, J6,6’ = 11.4 Hz, H-6'), 3.74 (d, 1H, J1,1 = 10.8 Hz, H-1'), 3.53 (d, 1H, J1,1 = 10.8 Hz, H-1'), 3.48 (dd, 1H, J2,1 = 3.5 Hz, J2,3 = 9.7 Hz, H-2), 3.33 (dd, 1H, J4,3 = 8.9 Hz, J4,5 = 10.1 Hz, H-4), 2.08 (s, 3H, CH3), 1.95 (s, 3H, CH3), 1.06 (s, 9H, t-Bu); 13C NMR (CDCl3) δ 170.3 (C=O), 170.2 (C=O), 138.5, 138.3, 138.3, 138.2, 137.8, 137.7, 135.6, 135.5, 133.2, 132.7, 129.7, 129.6, 128.5–127.5 (Ar), 104.6 (C-2'), 89.1 (C-1), 83.7 (C-3'), 81.6 (C-3), 81.3 (C-4'), 80.8 (C-5'), 79.8 (C-2), 76.9 (C-4), 75.6 (PhCH2), 75.0 (PhCH2), 73.5 (PhCH2), 73.3 (PhCH2), 72.8 (PhCH2), 71.8 (PhCH2), 71.4 (C-1'), 70.2 (C-5), 69.0 (C-6), 64.8 (C-7), 63.7 (C-6'), 26.9, 21.0, 20.8, 19.2; anal. calcd for C75H82O14Si × ½H2O (1253.57): C, 72.38; H, 6.72; found: C, 72.34; H, 6.29. HRMS (ESI) calc. for C75H86NO14Si [M + NH4]+: 1252.5818; found: 1252.5819.
Supporting Information
| Supporting Information File 1: The 1H and 13C NMR spectra of all new compounds (9–16Ac). | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737
Return to citation in text: [1] -
Rapi, Z.; Démuth, B.; Keglevich, G.; Grün, A.; Drahos, L.; Sóti, P. L.; Bakó, P. Tetrahedron: Asymmetry 2014, 25, 141–147. doi:10.1016/j.tetasy.2013.12.007
Return to citation in text: [1] -
Zhang, X. X.; Bradshaw, J. S.; Izatt, R. M. Chem. Rev. 1997, 97, 3313–3362. doi:10.1021/cr960144p
Return to citation in text: [1] -
Cheng, C.; Cai, Z.; Peng, X.-S.; Wong, H. N. C. J. Org. Chem. 2013, 78, 8562–8573. doi:10.1021/jo401240k
Return to citation in text: [1] -
Jarosz, S.; Listkowski, A. Curr. Org. Chem. 2006, 10, 643–662. doi:10.2174/138527206776359702
Return to citation in text: [1] -
Bako, P.; Keglevich, G.; Rapi, Z.; Toke, L. Curr. Org. Chem. 2012, 16, 297–304. doi:10.2174/138527212799499877
Return to citation in text: [1] -
Jarosz, S.; Potopnyk, M. A.; Kowalski, M. Sucrose as chiral platform in the synthesis of macrocyclic receptors. In Carbohydrate Chemistry-Chemical and Biological Approaches; Rauter, A. P.; Lindhorst, T.; Queneau, Y., Eds.; RSC Publishing, 2014; Vol. 40, pp 236–256. doi:10.1039/9781849739986-00236
See for a recent review.
Return to citation in text: [1] -
Jarosz, S.; Listkowski, A. Can. J. Chem. 2006, 84, 492–496. doi:10.1139/v06-035
Return to citation in text: [1] [2] -
Jarosz, S.; Listkowski, A.; Lewandowski, B. Phosphorus, Sulfur Silicon Relat. Elem. 2009, 184, 1285–1295. doi:10.1080/10426500902856370
Return to citation in text: [1] -
Queneau, Y.; Jarosz, S.; Lewandowski, B.; Fitremann, J. Adv. Carbohydr. Chem. Biochem. 2007, 61, 217–292. doi:10.1016/S0065-2318(07)61005-1
Return to citation in text: [1] -
Jarosz, S.; Lewandowski, B. Carbohydr. Res. 2008, 343, 965–969. doi:10.1016/j.carres.2008.01.016
Return to citation in text: [1] -
Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003
Return to citation in text: [1] -
Lewandowski, B.; Jarosz, S. Chem. Commun. 2008, 6399–6401. doi:10.1039/b816476b
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427
Return to citation in text: [1] -
Lewandowski, B.; Jarosz, S. Org. Lett. 2010, 12, 2532–2535. doi:10.1021/ol100749m
Return to citation in text: [1] -
Jarosz, S. J. Carbohydr. Chem. 2001, 20, 93–107. doi:10.1081/CAR-100103951
Return to citation in text: [1] -
Jarosz, S.; Mach, M. J. Chem. Soc., Perkin Trans. 1 1998, 3943–3948. doi:10.1039/a807190j
Return to citation in text: [1] -
Jarosz, S. Curr. Org. Chem. 2008, 12, 985–994. doi:10.2174/138527208785161187
Return to citation in text: [1] -
Jarosz, S.; Mach, M.; Frelek, J. J. Carbohydr. Chem. 2000, 19, 693–715. doi:10.1080/07328300008544111
Return to citation in text: [1] -
Mach, M.; Jarosz, S. J. Carbohydr. Chem. 2001, 20, 411–424. doi:10.1081/CAR-100105713
Return to citation in text: [1] -
Synthesis of this aldehyde was already reported by us as intermediate in the preparation of the uronate 8 (see ref. [8]).
Return to citation in text: [1] -
Makosza, M.; Fedorynski, M. Adv. Catal. 1987, 35, 375–422. doi:10.1016/S0360-0564(08)60097-8
Return to citation in text: [1] -
Makosza, M. Pure Appl. Chem. 2000, 72, 1399–1403. doi:10.1351/pac200072071399
Return to citation in text: [1] -
Makosza, M.; Fedoryński, M. Catal. Rev.: Sci. Eng. 2003, 45, 321–367. doi:10.1081/CR-120025537
Return to citation in text: [1] -
Jarosz, S. Carbohydr. Res. 1988, 183, 201–207. doi:10.1016/0008-6215(88)84074-6
Return to citation in text: [1] -
Cha, J. K.; Christ, W. J.; Kishi, Y. Tetrahedron 1984, 40, 2247–2255. doi:10.1016/0040-4020(84)80008-3
Return to citation in text: [1] -
Frelek, J.; Pakulski, Z.; Zamojski, A. Tetrahedron: Asymmetry 1996, 7, 1363–1372. doi:10.1016/0957-4166(96)00153-X
Return to citation in text: [1] -
Frelek, J.; Ikekawa, N.; Takatsuto, S.; Snatzke, G. Chirality 1997, 9, 578–582. doi:10.1002/(SICI)1520-636X(1997)9:5/6<578::AID-CHIR27>3.0.CO;2-K
Return to citation in text: [1] -
Frelek, J.; Klimek, A.; Ruśkowska, P. Curr. Org. Chem. 2003, 7, 1081–1104. doi:10.2174/1385272033486576
Return to citation in text: [1] -
Jawiczuk, M.; Górecki, M.; Suszczyńska, A.; Karchier, M.; Jaźwiński, J.; Frelek, J. Inorg. Chem. 2013, 52, 8250–8263. doi:10.1021/ic401170m
Return to citation in text: [1] -
Biela, A.; Oulaïdi, F.; Gallienne, E.; Górecki, M.; Frelek, J.; Martin, O. R. Tetrahedron 2013, 69, 3348–3354. doi:10.1016/j.tet.2012.12.082
Return to citation in text: [1] -
Schönemann, W.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Nakagawa, S.; Kato, A.; Adachi, I.; Górecki, M.; Frelek, J.; Martin, O. R. ChemMedChem 2013, 8, 1805–1817. doi:10.1002/cmdc.201300327
Return to citation in text: [1]
| 30. | Jawiczuk, M.; Górecki, M.; Suszczyńska, A.; Karchier, M.; Jaźwiński, J.; Frelek, J. Inorg. Chem. 2013, 52, 8250–8263. doi:10.1021/ic401170m |
| 31. | Biela, A.; Oulaïdi, F.; Gallienne, E.; Górecki, M.; Frelek, J.; Martin, O. R. Tetrahedron 2013, 69, 3348–3354. doi:10.1016/j.tet.2012.12.082 |
| 32. | Schönemann, W.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Nakagawa, S.; Kato, A.; Adachi, I.; Górecki, M.; Frelek, J.; Martin, O. R. ChemMedChem 2013, 8, 1805–1817. doi:10.1002/cmdc.201300327 |
| 8. | Jarosz, S.; Listkowski, A. Can. J. Chem. 2006, 84, 492–496. doi:10.1139/v06-035 |
| 1. | Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737 |
| 2. | Rapi, Z.; Démuth, B.; Keglevich, G.; Grün, A.; Drahos, L.; Sóti, P. L.; Bakó, P. Tetrahedron: Asymmetry 2014, 25, 141–147. doi:10.1016/j.tetasy.2013.12.007 |
| 8. | Jarosz, S.; Listkowski, A. Can. J. Chem. 2006, 84, 492–496. doi:10.1139/v06-035 |
| 26. | Cha, J. K.; Christ, W. J.; Kishi, Y. Tetrahedron 1984, 40, 2247–2255. doi:10.1016/0040-4020(84)80008-3 |
| 7. |
Jarosz, S.; Potopnyk, M. A.; Kowalski, M. Sucrose as chiral platform in the synthesis of macrocyclic receptors. In Carbohydrate Chemistry-Chemical and Biological Approaches; Rauter, A. P.; Lindhorst, T.; Queneau, Y., Eds.; RSC Publishing, 2014; Vol. 40, pp 236–256. doi:10.1039/9781849739986-00236
See for a recent review. |
| 27. | Frelek, J.; Pakulski, Z.; Zamojski, A. Tetrahedron: Asymmetry 1996, 7, 1363–1372. doi:10.1016/0957-4166(96)00153-X |
| 28. | Frelek, J.; Ikekawa, N.; Takatsuto, S.; Snatzke, G. Chirality 1997, 9, 578–582. doi:10.1002/(SICI)1520-636X(1997)9:5/6<578::AID-CHIR27>3.0.CO;2-K |
| 29. | Frelek, J.; Klimek, A.; Ruśkowska, P. Curr. Org. Chem. 2003, 7, 1081–1104. doi:10.2174/1385272033486576 |
| 5. | Jarosz, S.; Listkowski, A. Curr. Org. Chem. 2006, 10, 643–662. doi:10.2174/138527206776359702 |
| 6. | Bako, P.; Keglevich, G.; Rapi, Z.; Toke, L. Curr. Org. Chem. 2012, 16, 297–304. doi:10.2174/138527212799499877 |
| 22. | Makosza, M.; Fedorynski, M. Adv. Catal. 1987, 35, 375–422. doi:10.1016/S0360-0564(08)60097-8 |
| 23. | Makosza, M. Pure Appl. Chem. 2000, 72, 1399–1403. doi:10.1351/pac200072071399 |
| 24. | Makosza, M.; Fedoryński, M. Catal. Rev.: Sci. Eng. 2003, 45, 321–367. doi:10.1081/CR-120025537 |
| 3. | Zhang, X. X.; Bradshaw, J. S.; Izatt, R. M. Chem. Rev. 1997, 97, 3313–3362. doi:10.1021/cr960144p |
| 4. | Cheng, C.; Cai, Z.; Peng, X.-S.; Wong, H. N. C. J. Org. Chem. 2013, 78, 8562–8573. doi:10.1021/jo401240k |
| 25. | Jarosz, S. Carbohydr. Res. 1988, 183, 201–207. doi:10.1016/0008-6215(88)84074-6 |
| 16. | Jarosz, S. J. Carbohydr. Chem. 2001, 20, 93–107. doi:10.1081/CAR-100103951 |
| 17. | Jarosz, S.; Mach, M. J. Chem. Soc., Perkin Trans. 1 1998, 3943–3948. doi:10.1039/a807190j |
| 18. | Jarosz, S. Curr. Org. Chem. 2008, 12, 985–994. doi:10.2174/138527208785161187 |
| 8. | Jarosz, S.; Listkowski, A. Can. J. Chem. 2006, 84, 492–496. doi:10.1139/v06-035 |
| 15. | Lewandowski, B.; Jarosz, S. Org. Lett. 2010, 12, 2532–2535. doi:10.1021/ol100749m |
| 21. | Synthesis of this aldehyde was already reported by us as intermediate in the preparation of the uronate 8 (see ref. [8]). |
| 10. | Queneau, Y.; Jarosz, S.; Lewandowski, B.; Fitremann, J. Adv. Carbohydr. Chem. Biochem. 2007, 61, 217–292. doi:10.1016/S0065-2318(07)61005-1 |
| 11. | Jarosz, S.; Lewandowski, B. Carbohydr. Res. 2008, 343, 965–969. doi:10.1016/j.carres.2008.01.016 |
| 12. | Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003 |
| 13. | Lewandowski, B.; Jarosz, S. Chem. Commun. 2008, 6399–6401. doi:10.1039/b816476b |
| 14. | Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427 |
| 9. | Jarosz, S.; Listkowski, A.; Lewandowski, B. Phosphorus, Sulfur Silicon Relat. Elem. 2009, 184, 1285–1295. doi:10.1080/10426500902856370 |
| 19. | Jarosz, S.; Mach, M.; Frelek, J. J. Carbohydr. Chem. 2000, 19, 693–715. doi:10.1080/07328300008544111 |
| 20. | Mach, M.; Jarosz, S. J. Carbohydr. Chem. 2001, 20, 411–424. doi:10.1081/CAR-100105713 |
© 2014 Pakulski et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)