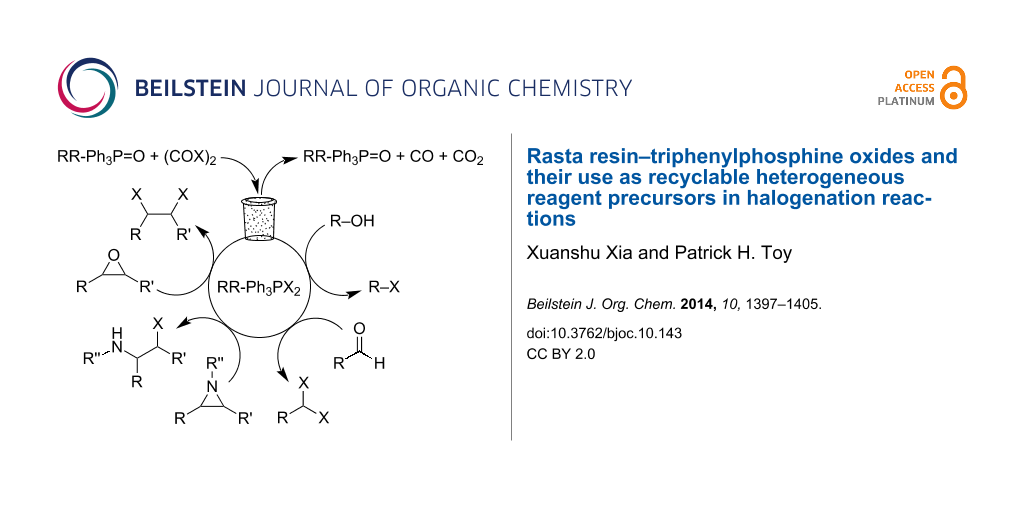
Abstract
Heterogeneous polymer-supported triphenylphosphine oxides based on the rasta resin architecture have been synthesized, and applied as reagent precursors in a wide range of halogenation reactions. The rasta resin–triphenylphosphine oxides were reacted with either oxalyl chloride or oxalyl bromide to form the corresponding halophosphonium salts, and these in turn were reacted with alcohols, aldehydes, aziridines and epoxides to form halogenated products in high yields after simple purification. The polymer-supported triphenylphosphine oxides formed as a byproduct during these reactions could be recovered and reused numerous times with no appreciable decrease in reactivity.

Graphical Abstract
Introduction
One of the major drawbacks of the Wittig [1] and Mitsunobu [2,3] reactions is that they result in the formation of a stoichiometric quantity of triphenylphosphine oxide (1) as a byproduct. From an atom economy perspective this is less than ideal, and from an environmental point of view it would be good if 1 could be simply reduced to triphenylphosphine (2) for reuse [4]. In this regard Tanaka and co-workers have studied the possibility of applying the reaction first reported by Masaki and Fukui [5] in which 1 can be treated with oxalyl chloride (or bromide) to form halophosphonium salt 3a (or 3b), which in turn can be reduced to 2 under more mild reaction conditions than can 1 (Scheme 1) [6,7].
![[1860-5397-10-143-i1]](/bjoc/content/inline/1860-5397-10-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Masaki–Fukui reaction and halophosphonium salt reduction.
Scheme 1: The Masaki–Fukui reaction and halophosphonium salt reduction.
In addition to being relatively easily reduced, halophosphonium salts 3a,b are also useful reagents in a wide range of reactions, such as those illustrated in Scheme 2: (1) the conversion of alcohols 4 to alkyl halides 5 (the Appel reaction), (2) the conversion of aldehydes 6 to 1,1-dihaloalkanes 7, (3) halogenation of aziridines 8 to form 2-haloamines 9, (4) halogenation of epoxides 10 to form 1,2-dihaloalkanes 11, (5) and the dehydration of oximes 12 to form nitriles 13.
![[1860-5397-10-143-i2]](/bjoc/content/inline/1860-5397-10-143-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Representative reactions involving halophosphonium salts 3a,b.
Scheme 2: Representative reactions involving halophosphonium salts 3a,b.
Capitalizing on the fact that 1 is formed as a byproduct from 3a,b in each of these reactions, Denton and co-workers have recently combined the Masaki–Fukui reaction with many of the functional group transformation outlined in Scheme 2, in one-pot processes in which the role of 1 is referred to as that of a catalyst [8-12]. For example, catalytic Appel reactions were achieved by slowly adding separate solutions of oxalyl chloride and alcohols 4 to a solution of 1 (Scheme 3) [8,9]. In these reactions, the simultaneous slow addition of oxalyl chloride and alcohol substrate 4 to a sub-stoichiometric quantity of 3a was necessary in order to minimize formation of undesired ester side-products formed by the reaction of 4 with the acid chloride. Furthermore, chromatographic purification of the alkyl halide product 5 was required. Thus, while the procedures reported by Denton et al. might be conceptually interesting, they may not be particularly convenient to perform, especially on larger scales than what was originally reported.
![[1860-5397-10-143-i3]](/bjoc/content/inline/1860-5397-10-143-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Catalytic Appel reactions reported by Denton and co-workers.
Scheme 3: Catalytic Appel reactions reported by Denton and co-workers.
We have had a long-term interest in the use of organic polymers as supports for reagents and catalysts [13], and have reported the use of various polymer-supported phosphines as reagents, organocatalysts, and ligands in order to simplify product isolation [14-18]. Most recently we have studied the use of the rasta resin polystyrene architecture [19-26] as a platform for reagents and catalysts [27-33], and have used easily synthesized rasta resin–Ph3P (14) in various Wittig reactions that required only filtration and solvent removal for product purification (Figure 1) [27-29]. Additionally, 14 was converted into phosphonium salt 15, which proved to be an efficient and highly recyclable catalyst for aldehyde and ketone cyanosilylation reactions from which the products could also be obtained pure after only filtration and solvent removal [30]. It should be noted that the grafts of the rasta resins reported are random co-polymers, and the structures drawn for them are not mean to indicate that they are block co-polymers. The format for their presentation is used merely to indicate their monomer incorporation ratios.
![[1860-5397-10-143-1]](/bjoc/content/figures/1860-5397-10-143-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Thus, considering our prior success in using 14 and 15, we wanted to oxidize 14 to 16, and in turn use this as a heterogeneous precursor to reagents 17a,b for use in the halogenation reactions described in Scheme 2. We anticipated that using a full equivalent 17a,b generated in situ would eliminate the need for slow addition of the oxalyl halide to form the halophosphonium salt, and thus allow for the reactions to be performed more conveniently than in the catalytic procedures of Denton and co-workers. Furthermore, since 16 would be the byproduct of the reactions, it could be recovered by filtration at the end of the reactions and reused directly. Herein we report the realization of this strategy and describe simple procedures for alcohol, aziridine, aldehyde and epoxide halogenation reactions from which the desired products are easily isolated and the phosphine oxide byproduct is readily recycled.
Results and Discussion
Rasta resin 16 was prepared by oxidation of 14, which was prepared as previously reported [28], using H2O2 (Scheme 4). The loading level of 16 was determined by elemental analysis to be 0.97 mmol/g, and gel-phase 31P NMR spectroscopic analysis of 16 showed only a single peak at 29.4 ppm, indicating that the phosphine groups were completely oxidized.
![[1860-5397-10-143-i4]](/bjoc/content/inline/1860-5397-10-143-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis and applications of rasta resins 16 and 17a,b.
Scheme 4: Synthesis and applications of rasta resins 16 and 17a,b.
Appel reactions using 16
With 16 in hand, we initially used it to perform Appel reactions by first converting it into either 17a or 17b in situ (Scheme 4). To do this, 16 was suspended/swollen in dichloromethane, and then the appropriate oxalyl halide was added. Once gas evolution ceased, alcohol 4 was added, and the reaction mixture was heated to reflux. Progress of the reactions was monitored by TLC analysis, and they were all finished in 4–7 hours. Upon completion, the reaction mixtures were cooled to room temperature and then filtered. Finally, the filtrates were concentrated at reduced pressure to afford the desired products that were essentially pure based on 1H and 13C NMR spectroscopic analyses. Chromatographic purification of the resultant alkyl halide products was not required. As can be seen in Table 1, both primary (entries 1–8) and secondary (entries 9 and 10) benzylic alcohols with various substituents all afforded excellent yield of the corresponding chloride and bromide using this procedure. Similar high yields were also obtained from reactions using primary (Table 1, entries 11–14) and secondary aliphatic alcohols (Table 1, entries 15 and 16). A reaction performed on a scale ten times larger afforded excellent yield as well (Table 1, entry 17). The recovered polymer was washed sequentially with water, MeOH, THF, diethyl ether and hexane. After drying, 31P NMR spectroscopy confirmed its identity as 16.
Table 1: Appel reactions using 16.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-143-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting material | Product | Yield (%) | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-143-i7.svg?max-width=637&scale=1.0)
|
4A |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-143-i8.svg?max-width=637&scale=1.0)
|
5Aa, X = Cl | 98 |
| 2 | 5Ab, X = Br | 92 | |||
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-143-i9.svg?max-width=637&scale=1.0)
|
4B |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-143-i10.svg?max-width=637&scale=1.0)
|
5Ba, X = Cl | 93 |
| 4 | 5Bb, X = Br | 91 | |||
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-143-i11.svg?max-width=637&scale=1.0)
|
4C |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-143-i12.svg?max-width=637&scale=1.0)
|
5Ca, X = Cl | 93 |
| 6 | 5Cb, X = Br | 90 | |||
| 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-143-i13.svg?max-width=637&scale=1.0)
|
4D |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-143-i14.svg?max-width=637&scale=1.0)
|
5Da, X = Cl | 98 |
| 8 | 5Db, X = Br | 93 | |||
| 9 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-143-i15.svg?max-width=637&scale=1.0)
|
4E |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-143-i16.svg?max-width=637&scale=1.0)
|
5Ea, X = Cl | 85 |
| 10 | 5Eb, X = Br | 89 | |||
| 11 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-143-i17.svg?max-width=637&scale=1.0)
|
4F |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-143-i18.svg?max-width=637&scale=1.0)
|
5Fa, X = Cl | 95 |
| 12 | 5Fb, X = Br | 90 | |||
| 13 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-143-i19.svg?max-width=637&scale=1.0)
|
4G |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-143-i20.svg?max-width=637&scale=1.0)
|
5Ga, X = Cl | 98 |
| 14 | 5Gb, X = Br | 92 | |||
| 15 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-143-i21.svg?max-width=637&scale=1.0)
|
4H |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-143-i22.svg?max-width=637&scale=1.0)
|
5Ha, X = Cl | 95 |
| 16 | 5Hb, X = Br | 91 | |||
| 17b | 5Ha, X = Cl | 92 | |||
aReaction conditions: 0.6 mmol 16, 0.6 mmol oxalyl halide, 0.5 mmol 4 in 5 mL CH2Cl2, reflux. bReaction conditions: 6 mmol 16, 6 mmol oxalyl halide, 5 mmol 4 in 50 mL CH2Cl2, reflux.
Aldehyde halogenation reactions using 16
With the success of the Appel reactions, we further examined the utility of 16 by studying its use in aldehyde halogenation reactions. As before, 16 was converted into 17a or 17b in situ, and aldehyde 6 was added upon cessation of gas evolution. After 72 hours at reflux, the reactions were stopped, filtered and the products were purified by column chromatography (Table 2). Unlike the Appel reactions discussed above, some of these reactions did not proceed to completion, even when the reaction time was lengthened. Generally, it was observed that the bromination reactions afforded higher product yields than did the corresponding chlorination reactions (Table 2, entries 1–8), except when electron-rich aldehyde starting materials were used (Table 2, entries 9–12). These results are generally similar to what was previously reported by Denton and co-workers when similar substrates were used [12].
Table 2: Halogenation reactions of aldehydes using 16.a
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-143-i23.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting material | Product | Yield (%) | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-143-i24.svg?max-width=637&scale=1.0)
|
6A |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-143-i25.svg?max-width=637&scale=1.0)
|
7Aa, X = Cl | 54b |
| 2 | 7Ab, X = Br | 75b | |||
| 3 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-143-i26.svg?max-width=637&scale=1.0)
|
6B |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-143-i27.svg?max-width=637&scale=1.0)
|
7Ba, X = Cl | 65b |
| 4 | 7Bb, X = Br | 83b | |||
| 5 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-143-i28.svg?max-width=637&scale=1.0)
|
6C |
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-143-i29.svg?max-width=637&scale=1.0)
|
7Ca, X = Cl | 64b |
| 6 | 7Cb, X = Br | 89b | |||
| 7 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-10-143-i30.svg?max-width=637&scale=1.0)
|
6D |
![[Graphic 26]](/bjoc/content/inline/1860-5397-10-143-i31.svg?max-width=637&scale=1.0)
|
7Da, X = Cl | 61b |
| 8 | 7Db, X = Br | 93b | |||
| 9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-10-143-i32.svg?max-width=637&scale=1.0)
|
6E |
![[Graphic 28]](/bjoc/content/inline/1860-5397-10-143-i33.svg?max-width=637&scale=1.0)
|
7Ea, X = Cl | 94c |
| 10 | 7Eb, X = Br | 20d | |||
| 11 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-10-143-i34.svg?max-width=637&scale=1.0)
|
6F |
![[Graphic 30]](/bjoc/content/inline/1860-5397-10-143-i35.svg?max-width=637&scale=1.0)
|
7Fa, X = Cl | 85b |
| 12 | 7Fb. X = Br | 77b | |||
aReaction conditions: 0.3 mmol 16, 0.3 mmol oxalyl halide, 0.1 mmol 6 in 3 mL CHCl3, reflux. bIsolated yield after flash silica gel column chromatography. cIsolated yield after filtration and concentration under reduced pressure. dDetermined by 1H NMR spectroscopy.
Aziridine halogenation reactions using 16
We next examined the use of 16 as a precursor to 17a and 17b in aziridine halogenation reactions [34]. Using reaction conditions similar to those used for the Appel and aldehyde halogenation reactions, a variety of N-tosyl aziridines 8 were successfully converted into the corresponding chloro- or bromotosylamides 9 in excellent yields (Table 3). The trans configurations of the 9Ba and 9Bb were confirmed by X-ray diffraction analysis of the isolated products, and as was true for the Appel reactions described above, all the products were obtained in high purity simply by filtration to remove the polymer, and concentration of the filtrate. Our procedure was also successfully performed on a gram-scale (Table 3, entry 5), and substrates 8 possessing a single aryl substituent were halogenated as the less hindered position (Table 3, entries 10–13).
Table 3: Halogenation reactions of aziridines using 16.a
![[Graphic 31]](/bjoc/content/inline/1860-5397-10-143-i36.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting material | Product | Yield (%) | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-10-143-i37.svg?max-width=637&scale=1.0)
|
8A |
![[Graphic 33]](/bjoc/content/inline/1860-5397-10-143-i38.svg?max-width=637&scale=1.0)
|
9Aa, X = Cl | 95 |
| 2 | 9Ab, X = Br | 93 | |||
| 3 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-10-143-i39.svg?max-width=637&scale=1.0)
|
8B |
![[Graphic 35]](/bjoc/content/inline/1860-5397-10-143-i40.svg?max-width=637&scale=1.0)
|
9Ba, X = Cl | 89 |
| 4 | 9Bb, X = Br | 92 | |||
| 5b | 9Ba, X = Cl | 95 | |||
| 6 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-10-143-i41.svg?max-width=637&scale=1.0)
|
8C |
![[Graphic 37]](/bjoc/content/inline/1860-5397-10-143-i42.svg?max-width=637&scale=1.0)
|
9Ca, X = Cl | 96 |
| 7 | 9Cb, X = Br | 98 | |||
| 8 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-10-143-i43.svg?max-width=637&scale=1.0)
|
8D |
![[Graphic 39]](/bjoc/content/inline/1860-5397-10-143-i44.svg?max-width=637&scale=1.0)
|
9Da, X = Cl | 93 |
| 9 | 9Db, X = Br | 92 | |||
| 10 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-10-143-i45.svg?max-width=637&scale=1.0)
|
8E |
![[Graphic 41]](/bjoc/content/inline/1860-5397-10-143-i46.svg?max-width=637&scale=1.0)
|
9Ea, X = Cl | 91 |
| 11 | 9Eb, X = Br | 93 | |||
| 12 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-10-143-i47.svg?max-width=637&scale=1.0)
|
8F |
![[Graphic 43]](/bjoc/content/inline/1860-5397-10-143-i48.svg?max-width=637&scale=1.0)
|
9Fa, X = Cl | 93 |
| 13 | 9Fb, X = Br | 95 | |||
aReaction conditions: 0.6 mmol 16, 0.6 mmol oxalyl halide, 0.5 mmol 8 in 5 mL CH2Cl2, reflux. bReaction conditions: 6 mmol 16, 6 mmol oxalyl halide, 5 mmol 8 in 50 mL CH2Cl2, reflux.
Recovery and reuse of 16
After demonstrating that 16 could effectively serve as a precursor to 17a,b, and realizing that it was returned as the byproduct of the above reactions, we next examined its recyclability in the chlorination of alcohol 4H and aziridine 8B. The results of these studies are shown in Table 4 and Table 5, respectively. In these experiments, the polymer recovered by filtration of the reaction mixture was washed and dried, and then used directly for the next reaction cycle. Excellent yields were successfully obtained for 8 runs with both 4H and 8B. Gel-phase 31P NMR analysis of 16 recovered at the end of these experiments indicated no change in its oxidation state.
Table 4: Recycling of 16 in the chlorination of 4H.a
![[Graphic 44]](/bjoc/content/inline/1860-5397-10-143-i49.svg?max-width=637&scale=1.0)
|
||||||||
| Run | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Yield of 5Ha (%) | 94 | 95 | 93 | 98 | 94 | 95 | 95 | 93 |
| Recovery of 16 (%) | 99 | 99 | 99 | 99 | 98 | 95 | 93 | 91 |
aReaction conditions: 0.6 mmol 16, 0.6 mmol oxalyl chloride, 0.5 mmol 4H in 5 mL CH2Cl2, reflux.
Table 5: Recycling of 16 in the chlorination of 8B.a
![[Graphic 45]](/bjoc/content/inline/1860-5397-10-143-i50.svg?max-width=637&scale=1.0)
|
||||||||
| Run | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Yield of 9Ba (%) | 93 | 92 | 95 | 93 | 87 | 91 | 91 | 90 |
| Recovery of 16 (%) | 96 | 97 | 95 | 95 | 96 | 95 | 90 | 92 |
aReaction conditions: 0.6 mmol 16, 0.6 mmol oxalyl chloride, 0.5 mmol 8B in 5 mL CH2Cl2, reflux.
Epoxide halogenation reactions
With the versatility and excellent reactivity of 16 established, we were encouraged to examine our method in the epoxide halogenation reactions shown in Scheme 2. Since these reactions require the use of a base, we designed a bifunctional rasta resin, RR-NBniPr2-PPh3=O 18 (Scheme 5), which bears both triphenylphosphine oxide and tertiary amine moieties, in order to increase the efficiency and appeal of our method. We have extensive experience in preparing functionalized resins with two different catalytic groups [35-38], and prepared 18 by oxidation of 19, which we previously used as a bifunctional reagent in one-pot Wittig reactions [29]. Gel-phase 31P NMR spectroscopic analysis of 18 indicated that oxidation of the phosphine groups was complete, and elemental analysis was used to determine the loading level of phosphine oxide and amine groups to be 1.07 mmol/g and 1.06 mmol/g, respectively. It should be noted that a test reaction between N,N-diisopropylbenzylamine and H2O2 under similar reaction conditions does not result in amine oxidation, and this seems to indicate that only the phosphine groups of 19 are oxidized during its conversion to 18.
![[1860-5397-10-143-i5]](/bjoc/content/inline/1860-5397-10-143-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of bifuncitonal rasta resin 18.
Scheme 5: Synthesis of bifuncitonal rasta resin 18.
Having successfully synthesized polymer 18, we examined its reactivity in epoxide halogenation reactions (Table 6). As before, 18 was suspended/swollen in solvent prior to addition of the oxalyl halide. Once gas evolution ceased, epoxide 10 was added, and the reaction mixture was heated to reflux. When TLC analysis indicated that the reactions were complete, 3–4 hours, the reaction mixtures were filtered and the filtrates were concentrated to afford products that were essentially pure according to both 1H and13C NMR analysis. Reactions with epoxides bearing phenyl (Table 6, entries 1 and 2), benzyl (Table 6, entries 3 and 4), and alkyl substutients (Table 6, entries 5–8) all proceeded to completion, and afforded the corresponding 1,2-dihalides in excellent yields.
Table 6: Halogenation reactions of epoxides using 18.a
![[Graphic 46]](/bjoc/content/inline/1860-5397-10-143-i51.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yield (%) | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-10-143-i52.svg?max-width=637&scale=1.0)
|
10A |
![[Graphic 48]](/bjoc/content/inline/1860-5397-10-143-i53.svg?max-width=637&scale=1.0)
|
11Aa, X = Cl | 95 |
| 2 | 11Ab, X = Br | 93 | |||
| 3 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-10-143-i54.svg?max-width=637&scale=1.0)
|
10B |
![[Graphic 50]](/bjoc/content/inline/1860-5397-10-143-i55.svg?max-width=637&scale=1.0)
|
11Ba, X = Cl | 89 |
| 4 | 11Bb, X = Br | 92 | |||
| 5 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-10-143-i56.svg?max-width=637&scale=1.0)
|
10C |
![[Graphic 52]](/bjoc/content/inline/1860-5397-10-143-i57.svg?max-width=637&scale=1.0)
|
11Ca, X = Cl | 95 |
| 6 | 11Cb, X = Br | 96 | |||
| 7 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-10-143-i58.svg?max-width=637&scale=1.0)
|
10D |
![[Graphic 54]](/bjoc/content/inline/1860-5397-10-143-i59.svg?max-width=637&scale=1.0)
|
11Da, X = Cl | 98 |
| 8 | 11Db, X = Br | 92 | |||
aReaction conditions: 1.2 mmol 18, 1.1 mmol oxalyl halide, 0.5 mmol 10 in 5 mL CHCl3, reflux.
We also examined the recyclability of 18 in the halogenation of epoxide 10C (Table 7). As was the case for 16, bifunctional polymer 18 could be repeatedly recovered and reused without an observable decrease in its reactivity. However, whereas 16 could be reused directly after recover, reusing 18 required washing it with an aqueous solution of Na2CO3 after each reaction.
![[Graphic 55]](/bjoc/content/inline/1860-5397-10-143-i60.svg?max-width=637&scale=1.0)
Conclusion
In summary, we have designed and synthesized recyclable heterogeneous rasta resin-supported triphenylphosphine oxide 16, and have applied it as a phosphonium halide salt precursor in a wide range of halogenation reactions from which it is readily recovered and reused. The reusability of this polymer was demonstrated by the fact that all of the reactions reported herein involving 16 were performed by repeatedly reusing a single batch of it. We also prepared bifunctional rasta resin 18 which bears amine groups in addition to the phosphine oxide moieties for use in epoxide halogenation reactions. The products of all of these reactions, except for the aldehyde halogenation reactions, can be easily separated from the phosphine oxide functionalized polymer simply by filtration, and isolated directly in high purity. Thus, the use of polymers 16 or 18 as reagent precursors represents a convenient alternative to the “phosphine oxide-catalyzed” methods of Denton and co-workers, which generally require slow addition of the oxalyl halide and chromatographic purification of the product. In order to assess the overall utility of 16 and 18, we are currently examining their applications as organocatalysts [39-43], and will report results of these studies in due course.
Experimental
General procedure for Appel reactions using 16: Polymer 16 (0.6 g, 0.6 mmol) was suspended in dichloromethane (5 mL), and the oxalyl halide (0.6 mmol) was added. Upon cessation of gas evolution, alcohol 4 (0.5 mmol) was added, and the reaction mixture magnetically stirred and heated to reflux. After the reaction was completed as monitored by TLC, the mixture was cooled to room temperature and filtered. The solid on funnel was washed with dichloromethane (10 mL × 3). The solvent of filtrate was removed under reduced pressure to afford the desired product 5 in an essentially pure state based on 1H and 13C NMR spectroscopic analyses.
General procedure for aldehyde halogenation reactions using 16: Polymer 16 (0.3 g, 0.3 mmol) was suspended in chloroform (3 mL), and the oxalyl halide was added (0.3 mmol). Upon cessation of gas evolution, aldehyde 6 (0.1 mmol) was added, and the reaction mixture was magnetically stirred and heated to reflux. After 72 hours, the reaction was cooled to room temperature and filtered. The solid on funnel was washed with dichloromethane (5 mL × 3). The solvent of filtrate was removed under reduced pressure. The resulting crude product 7 was purified by flash silica gel column chromatography using ethyl acetate and hexane as eluent mixture.
General procedure for aziridine halogenation reactions of using 16: Polymer 16 (0.6 g, 0.6 mmol) was suspended in dichloromethane (5 mL), and the oxalyl halide was added (0.6 mmol). Upon cessation of gas evolution, aziridine 8 (0.5 mmol) was added, and the reaction mixture was magnetically stirred and heated to reflux. After the reaction was completed as monitored by TLC, the mixture was cooled to room temperature and filtered. The solid on funnel was washed with dichloromethane (10 mL × 3). The solvent of filtrate was removed under reduced pressure to afford the desired product 9 in an essentially pure state based on 1H and 13C NMR spectroscopic analyses.
General procedure for epoxide halogenation reactions using 18: Polymer 18 (1.3 g, 1.2 mmol) was suspended in chloroform (10 mL) and the oxalyl halide was added (1.1 mmol). Upon cessation of gas evolution, epoxide 10 (0.5 mmol) was added, and the reaction was magnetically stirred and heated to reflux. After the reaction was completed as monitored by TLC, the mixture was cooled to room temperature and filtered. The solid on funnel was washed with dichloromethane (10 mL × 3). The solvent of filtrate was removed under reduced pressure to afford the desired product 11 in an essentially pure state based on 1H and 13C NMR spectroscopic analyses.
General procedure for recovery and reuse of 16 and 18: After being separated from the reaction mixture by filtration, the polymer, 16 or 18, was rinsed sequentially using deionized water (30 mL), dichloromethane (50 mL), MeOH (50 mL), THF (50 mL), diethyl ether (50 mL), hexane (50 mL). It was then dried under vacuum at 60 °C prior to use in the next reaction cycle. Furthermore, 18 was initially washed with a saturated aqueous solution of Na2CO3 in order to ensure that it was deprotonated and ready for use in the next reaction cycle.
Note Added in Proof
After the initial submission of our manuscript we became aware of a recent report by Denton and co-workers regarding similar work using polystyrene-supported halophosphonium salts in Appel and dehydration reactions [44]. This work utilized a commercially available polymer-supported phosphine oxide based on the Merrifield resin architecture, and it is noteworthy that the reactions reported in this manuscript required a 6-fold excess of the halophosphonium salt compared to the substrate. Use of our rasta resins 16 and 18 required only a 20 mol % excess.
Supporting Information
| Supporting Information File 1: Additional experimental details and characterization data of synthesized compounds. | ||
| Format: PDF | Size: 8.1 MB | Download |
References
-
Wittig, G.; Geissler, G. Liebigs Ann. Chem. 1953, 580, 44–57. doi:10.1002/jlac.19535800107
Return to citation in text: [1] -
Mitsunobu, O.; Yamada, M. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. doi:10.1246/bcsj.40.2380
Return to citation in text: [1] -
But, T. Y. S.; Toy, P. H. Chem.–Asian J. 2007, 2, 1340–1355. doi:10.1002/asia.200700182
Return to citation in text: [1] -
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. J. L.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c
Return to citation in text: [1] -
Masaki, M.; Fukui, K. Chem. Lett. 1977, 151–152. doi:10.1246/cl.1977.151
Return to citation in text: [1] -
Yano, T.; Hoshino, M.; Kuroboshi, M.; Tanaka, H. Synlett 2010, 801–803. doi:10.1055/s-0029-1219194
Return to citation in text: [1] -
Yano, T.; Kuroboshi, M.; Tanaka, H. Tetrahedron Lett. 2010, 51, 698–701. doi:10.1016/j.tetlet.2009.11.115
Return to citation in text: [1] -
Denton, R. M.; An, J.; Adeniran, B. Chem. Commun. 2010, 46, 3025–3027. doi:10.1039/C002825H
Return to citation in text: [1] [2] -
Denton, R. M.; An, J.; Adeniran, B.; Blake, A. J.; Lewis, W.; Poulton, A. M. J. Org. Chem. 2011, 76, 6749–6767. doi:10.1021/jo201085r
Return to citation in text: [1] [2] -
Denton, R. M.; Tang, X. P.; Przeslak, A. Org. Lett. 2010, 12, 4678–4681. doi:10.1021/ol102010h
Return to citation in text: [1] -
Denton, R. M.; An, J.; Lindovska, P.; Lewis, W. Tetrahedron 2012, 68, 2899–2905. doi:10.1016/j.tet.2012.01.067
Return to citation in text: [1] -
An, J.; Tang, X. P.; Moore, J.; Lewis, W.; Denton, R. M. Tetrahedron 2013, 69, 8769–8776. doi:10.1016/j.tet.2013.07.100
Return to citation in text: [1] [2] -
Lu, J.; Toy, P. H. Chem. Rev. 2009, 109, 815–838. doi:10.1021/cr8004444
Return to citation in text: [1] -
Choi, M. K. W.; He, H. S.; Toy, P. H. J. Org. Chem. 2003, 68, 9831–9834. doi:10.1021/jo035226+
Return to citation in text: [1] -
Zhao, L.-J.; He, H. S.; Shi, M.; Toy, P. H. J. Comb. Chem. 2004, 6, 680–683. doi:10.1021/cc049917a
Return to citation in text: [1] -
Harned, A. M.; He, H. S.; Toy, P. H.; Flynn, D. L.; Hanson, P. R. J. Am. Chem. Soc. 2005, 127, 52–53. doi:10.1021/ja045188r
Return to citation in text: [1] -
Zhao, L.-J.; Kwong, C. K.-W.; Shi, M.; Toy, P. H. Tetrahedron 2005, 62, 12026–12032. doi:10.1016/j.tet.2005.05.108
Return to citation in text: [1] -
He, H. S.; Yan, J. J.; Shen, R.; Zhuo, S.; Toy, P. H. Synlett 2006, 563–566. doi:10.1055/s-2006-933128
Return to citation in text: [1] -
Hodges, J. C.; Harikrishnan, L. S.; Ault-Justus, S. J. Comb. Chem. 2000, 2, 80–88. doi:10.1021/cc990067m
Return to citation in text: [1] -
Lindsley, C. W.; Hodges, J. C.; Filzen, G. F.; Watson, B. M.; Geyer, A. G. J. Comb. Chem. 2000, 2, 550–559. doi:10.1021/cc000037f
Return to citation in text: [1] -
McAlpine, S. R.; Lindsley, C. W.; Hodges, J. C.; Leonard, D. M.; Filzen, G. F. J. Comb. Chem. 2001, 3, 1–5. doi:10.1021/cc000068a
Return to citation in text: [1] -
Wisnoski, D. D.; Leister, W. H.; Strauss, K. A.; Zhao, Z.; Lindsley, C. W. Tetrahedron Lett. 2003, 44, 4321–4325. doi:10.1016/S0040-4039(03)00951-1
Return to citation in text: [1] -
Fournier, D.; Pascual, S.; Montembault, V.; Haddleton, D. M.; Fontaine, L. J. Comb. Chem. 2006, 8, 522–530. doi:10.1021/cc0600122
Return to citation in text: [1] -
Fournier, D.; Pascual, S.; Montembault, V.; Fontaine, L. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5316–5328. doi:10.1002/pola.21644
Return to citation in text: [1] -
Pawluczyk, J. M.; McClain, R. T.; Denicola, C.; Mulhearn, J. J.; Rudd, D. J.; Lindsley, C. W. Tetrahedron Lett. 2007, 48, 1497–1501. doi:10.1016/j.tetlet.2006.12.040
Return to citation in text: [1] -
Chen, G.; Tao, L.; Mantovani, G.; Geng, J.; Nystroem, D.; Haddleton, D. M. Macromolecules 2007, 40, 7513–7520. doi:10.1021/ma071362v
Return to citation in text: [1] -
Leung, P. S. W.; Teng, Y.; Toy, P. H. Synlett 2010, 1997–2001. doi:10.1055/s-0030-1258130
Return to citation in text: [1] [2] -
Leung, P. S. W.; Teng, Y.; Toy, P. H. Org. Lett. 2010, 12, 4996–4999. doi:10.1021/ol1021614
Return to citation in text: [1] [2] [3] -
Teng, Y.; Lu, J.; Toy, P. H. Chem.–Asian J. 2012, 7, 351–359. doi:10.1002/asia.201100721
Return to citation in text: [1] [2] [3] -
Teng, Y.; Toy, P. H. Synlett 2011, 551–554. doi:10.1055/s-0030-1259515
Return to citation in text: [1] [2] -
Lu, J.; Toy, P. H. Synlett 2011, 659–662. doi:10.1055/s-0030-1259690
Return to citation in text: [1] -
Diebold, C.; Becht, J.-M.; Lu, J.; Toy, P. H.; Le Drian, C. Eur. J. Org. Chem. 2012, 893–896. doi:10.1002/ejoc.201101075
Return to citation in text: [1] -
Yang, Y.-C.; Leung, D. Y. C.; Toy, P. H. Synlett 2013, 1870–1874. doi:10.1055/s-0033-1338963
Return to citation in text: [1] -
Kumar, M.; Pandey, S. K.; Gandhi, S.; Singh, V. K. Tetrahedron Lett. 2009, 50, 363–365. doi:10.1016/j.tetlet.2008.11.009
Return to citation in text: [1] -
Kwong, C. K.-W.; Huang, R.; Zhang, M.; Shi, M.; Toy, P. H. Chem.–Eur. J. 2007, 13, 2369–2376. doi:10.1002/chem.200601197
Return to citation in text: [1] -
Kwong, C. K.-W.; Fu, M. Y.; Law, H. C.-H.; Toy, P. H. Synlett 2010, 2617–2620. doi:10.1055/s-0030-1258576
Return to citation in text: [1] -
Lu, J.; Toy, P. H. Synlett 2011, 1723–1726. doi:10.1055/s-0030-1260808
Return to citation in text: [1] -
Lu, J.; Toy, P. H. Synlett 2011, 2985–2990. doi:10.1055/s-0031-1289900
Return to citation in text: [1] -
Sugiura, M.; Sato, N.; Kotani, S.; Nakajima, M. Chem. Commun. 2008, 4309–4311. doi:10.1039/B807529H
Return to citation in text: [1] -
Sugiura, M.; Sato, N.; Sonoda, Y.; Kotani, S.; Nakajima, M. Chem.–Asian J. 2010, 5, 478–481. doi:10.1002/asia.200900450
Return to citation in text: [1] -
Cao, J.-J.; Zhou, F.; Zhou, J. Angew. Chem., Int. Ed. 2010, 49, 4976–4980. doi:10.1002/anie.201000896
Return to citation in text: [1] -
Chen, L.; Shi, T.-D.; Zhou, J. Chem.–Asian J. 2013, 8, 556–559. doi:10.1002/asia.201201130
Return to citation in text: [1] -
Lu, J.; Toy, P. H. Chem.–Asian J. 2011, 6, 2251–2254. doi:10.1002/asia.201100296
Return to citation in text: [1] -
Tang, X.; An, J.; Denton, R. M. Tetrahedron Lett. 2014, 55, 799–802. doi:10.1016/j.tetlet.2013.11.098
Return to citation in text: [1]
| 39. | Sugiura, M.; Sato, N.; Kotani, S.; Nakajima, M. Chem. Commun. 2008, 4309–4311. doi:10.1039/B807529H |
| 40. | Sugiura, M.; Sato, N.; Sonoda, Y.; Kotani, S.; Nakajima, M. Chem.–Asian J. 2010, 5, 478–481. doi:10.1002/asia.200900450 |
| 41. | Cao, J.-J.; Zhou, F.; Zhou, J. Angew. Chem., Int. Ed. 2010, 49, 4976–4980. doi:10.1002/anie.201000896 |
| 42. | Chen, L.; Shi, T.-D.; Zhou, J. Chem.–Asian J. 2013, 8, 556–559. doi:10.1002/asia.201201130 |
| 43. | Lu, J.; Toy, P. H. Chem.–Asian J. 2011, 6, 2251–2254. doi:10.1002/asia.201100296 |
| 35. | Kwong, C. K.-W.; Huang, R.; Zhang, M.; Shi, M.; Toy, P. H. Chem.–Eur. J. 2007, 13, 2369–2376. doi:10.1002/chem.200601197 |
| 36. | Kwong, C. K.-W.; Fu, M. Y.; Law, H. C.-H.; Toy, P. H. Synlett 2010, 2617–2620. doi:10.1055/s-0030-1258576 |
| 37. | Lu, J.; Toy, P. H. Synlett 2011, 1723–1726. doi:10.1055/s-0030-1260808 |
| 38. | Lu, J.; Toy, P. H. Synlett 2011, 2985–2990. doi:10.1055/s-0031-1289900 |
| 29. | Teng, Y.; Lu, J.; Toy, P. H. Chem.–Asian J. 2012, 7, 351–359. doi:10.1002/asia.201100721 |
| 1. | Wittig, G.; Geissler, G. Liebigs Ann. Chem. 1953, 580, 44–57. doi:10.1002/jlac.19535800107 |
| 6. | Yano, T.; Hoshino, M.; Kuroboshi, M.; Tanaka, H. Synlett 2010, 801–803. doi:10.1055/s-0029-1219194 |
| 7. | Yano, T.; Kuroboshi, M.; Tanaka, H. Tetrahedron Lett. 2010, 51, 698–701. doi:10.1016/j.tetlet.2009.11.115 |
| 12. | An, J.; Tang, X. P.; Moore, J.; Lewis, W.; Denton, R. M. Tetrahedron 2013, 69, 8769–8776. doi:10.1016/j.tet.2013.07.100 |
| 34. | Kumar, M.; Pandey, S. K.; Gandhi, S.; Singh, V. K. Tetrahedron Lett. 2009, 50, 363–365. doi:10.1016/j.tetlet.2008.11.009 |
| 4. | Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. J. L.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c |
| 2. | Mitsunobu, O.; Yamada, M. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. doi:10.1246/bcsj.40.2380 |
| 3. | But, T. Y. S.; Toy, P. H. Chem.–Asian J. 2007, 2, 1340–1355. doi:10.1002/asia.200700182 |
| 28. | Leung, P. S. W.; Teng, Y.; Toy, P. H. Org. Lett. 2010, 12, 4996–4999. doi:10.1021/ol1021614 |
| 14. | Choi, M. K. W.; He, H. S.; Toy, P. H. J. Org. Chem. 2003, 68, 9831–9834. doi:10.1021/jo035226+ |
| 15. | Zhao, L.-J.; He, H. S.; Shi, M.; Toy, P. H. J. Comb. Chem. 2004, 6, 680–683. doi:10.1021/cc049917a |
| 16. | Harned, A. M.; He, H. S.; Toy, P. H.; Flynn, D. L.; Hanson, P. R. J. Am. Chem. Soc. 2005, 127, 52–53. doi:10.1021/ja045188r |
| 17. | Zhao, L.-J.; Kwong, C. K.-W.; Shi, M.; Toy, P. H. Tetrahedron 2005, 62, 12026–12032. doi:10.1016/j.tet.2005.05.108 |
| 18. | He, H. S.; Yan, J. J.; Shen, R.; Zhuo, S.; Toy, P. H. Synlett 2006, 563–566. doi:10.1055/s-2006-933128 |
| 27. | Leung, P. S. W.; Teng, Y.; Toy, P. H. Synlett 2010, 1997–2001. doi:10.1055/s-0030-1258130 |
| 28. | Leung, P. S. W.; Teng, Y.; Toy, P. H. Org. Lett. 2010, 12, 4996–4999. doi:10.1021/ol1021614 |
| 29. | Teng, Y.; Lu, J.; Toy, P. H. Chem.–Asian J. 2012, 7, 351–359. doi:10.1002/asia.201100721 |
| 30. | Teng, Y.; Toy, P. H. Synlett 2011, 551–554. doi:10.1055/s-0030-1259515 |
| 31. | Lu, J.; Toy, P. H. Synlett 2011, 659–662. doi:10.1055/s-0030-1259690 |
| 32. | Diebold, C.; Becht, J.-M.; Lu, J.; Toy, P. H.; Le Drian, C. Eur. J. Org. Chem. 2012, 893–896. doi:10.1002/ejoc.201101075 |
| 33. | Yang, Y.-C.; Leung, D. Y. C.; Toy, P. H. Synlett 2013, 1870–1874. doi:10.1055/s-0033-1338963 |
| 27. | Leung, P. S. W.; Teng, Y.; Toy, P. H. Synlett 2010, 1997–2001. doi:10.1055/s-0030-1258130 |
| 28. | Leung, P. S. W.; Teng, Y.; Toy, P. H. Org. Lett. 2010, 12, 4996–4999. doi:10.1021/ol1021614 |
| 29. | Teng, Y.; Lu, J.; Toy, P. H. Chem.–Asian J. 2012, 7, 351–359. doi:10.1002/asia.201100721 |
| 8. | Denton, R. M.; An, J.; Adeniran, B. Chem. Commun. 2010, 46, 3025–3027. doi:10.1039/C002825H |
| 9. | Denton, R. M.; An, J.; Adeniran, B.; Blake, A. J.; Lewis, W.; Poulton, A. M. J. Org. Chem. 2011, 76, 6749–6767. doi:10.1021/jo201085r |
| 44. | Tang, X.; An, J.; Denton, R. M. Tetrahedron Lett. 2014, 55, 799–802. doi:10.1016/j.tetlet.2013.11.098 |
| 8. | Denton, R. M.; An, J.; Adeniran, B. Chem. Commun. 2010, 46, 3025–3027. doi:10.1039/C002825H |
| 9. | Denton, R. M.; An, J.; Adeniran, B.; Blake, A. J.; Lewis, W.; Poulton, A. M. J. Org. Chem. 2011, 76, 6749–6767. doi:10.1021/jo201085r |
| 10. | Denton, R. M.; Tang, X. P.; Przeslak, A. Org. Lett. 2010, 12, 4678–4681. doi:10.1021/ol102010h |
| 11. | Denton, R. M.; An, J.; Lindovska, P.; Lewis, W. Tetrahedron 2012, 68, 2899–2905. doi:10.1016/j.tet.2012.01.067 |
| 12. | An, J.; Tang, X. P.; Moore, J.; Lewis, W.; Denton, R. M. Tetrahedron 2013, 69, 8769–8776. doi:10.1016/j.tet.2013.07.100 |
| 19. | Hodges, J. C.; Harikrishnan, L. S.; Ault-Justus, S. J. Comb. Chem. 2000, 2, 80–88. doi:10.1021/cc990067m |
| 20. | Lindsley, C. W.; Hodges, J. C.; Filzen, G. F.; Watson, B. M.; Geyer, A. G. J. Comb. Chem. 2000, 2, 550–559. doi:10.1021/cc000037f |
| 21. | McAlpine, S. R.; Lindsley, C. W.; Hodges, J. C.; Leonard, D. M.; Filzen, G. F. J. Comb. Chem. 2001, 3, 1–5. doi:10.1021/cc000068a |
| 22. | Wisnoski, D. D.; Leister, W. H.; Strauss, K. A.; Zhao, Z.; Lindsley, C. W. Tetrahedron Lett. 2003, 44, 4321–4325. doi:10.1016/S0040-4039(03)00951-1 |
| 23. | Fournier, D.; Pascual, S.; Montembault, V.; Haddleton, D. M.; Fontaine, L. J. Comb. Chem. 2006, 8, 522–530. doi:10.1021/cc0600122 |
| 24. | Fournier, D.; Pascual, S.; Montembault, V.; Fontaine, L. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5316–5328. doi:10.1002/pola.21644 |
| 25. | Pawluczyk, J. M.; McClain, R. T.; Denicola, C.; Mulhearn, J. J.; Rudd, D. J.; Lindsley, C. W. Tetrahedron Lett. 2007, 48, 1497–1501. doi:10.1016/j.tetlet.2006.12.040 |
| 26. | Chen, G.; Tao, L.; Mantovani, G.; Geng, J.; Nystroem, D.; Haddleton, D. M. Macromolecules 2007, 40, 7513–7520. doi:10.1021/ma071362v |
© 2014 Xia and Toy; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)