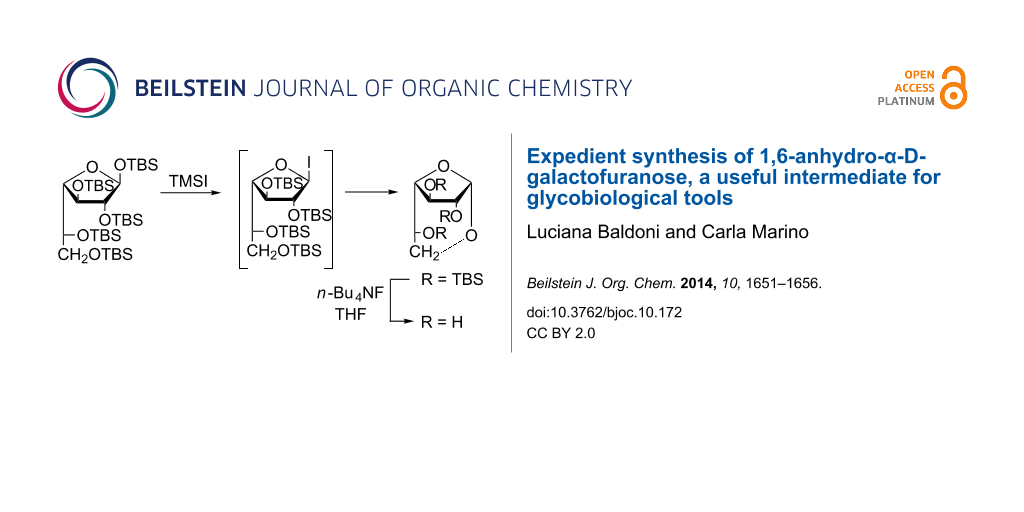
Expedient synthesis of 1,6-anhydro-α-D-galactofuranose, a useful intermediate for glycobiological tools
and
Luciana Baldoni
CIHIDECAR-CONICET-UBA, Departamento de Química Orgánica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón II, Ciudad Universitaria, 1428 Buenos Aires, Argentina, Tel/Fax: +54-11-45763352
CIHIDECAR-CONICET-UBA, Departamento de Química Orgánica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón II, Ciudad Universitaria, 1428 Buenos Aires, Argentina, Tel/Fax: +54-11-45763352
CIHIDECAR-CONICET-UBA, Departamento de Química Orgánica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón II, Ciudad Universitaria, 1428 Buenos Aires, Argentina, Tel/Fax: +54-11-45763352
Corresponding author email
Associate Editor: S. Flitsch Beilstein J. Org. Chem.2014,10, 1651–1656.https://doi.org/10.3762/bjoc.10.172 Received 24 Feb 2014,
Accepted 26 Jun 2014,
Published 21 Jul 2014
A new and efficient three-step procedure for the synthesis of 1,6-anhydro-α-D-galactofuranose is described. The key step involves the formation of the galactofuranosyl iodide by treatment of per-O-TBS-D-Galf with TMSI, the selective 6-O-desilylation by an excess of TMSI, and the simultaneous nucleophilic attack of the 6-hydroxy group on the anomeric carbon, with the iodide as a good leaving group. This compound is a good precursor for building blocks for the construction of 1→6 linkages.
Anhydro sugars are formed by the intramolecular elimination of a water molecule, with the simultaneous formation of a new heterocyclic ring of different size. They are valuable intermediates not only in carbohydrate synthesis, but also as starting materials for other natural and non-natural complex products and bioactive compounds. Among the glycosans, the anhydro sugars involving the anomeric center in the ring formation, the 1,6-anhydro sugars are the most common and useful building blocks [1,2]. They can play a role in synthetic methodologies aiming at the obtainment of regioselectively functionalized sugars in a few steps, which could give easy access to convenient glycosyl donors and acceptors [3].
Some sugars, for example galactose, can afford not only the pyranosic derivative 1 but also the furanosic 1,6-anhydro derivative 2, both of which may be equipped with [3.2.1] bicyclic skeletons (Figure 1) [1].
Figure 1:
Possible 1,6-anydro derivatives for D-galactose.
Figure 1:
Possible 1,6-anydro derivatives for D-galactose.
A variety of chemical approaches for the synthesis of 1,6-anhydro sugars have been developed [3-10]. Two classes of methods for the synthesis of 2 can be discriminated, the first of which starts from free galactose (D-Gal) and afford mixtures of 1 and 2 and the second starts from a galactofuranose (D-Galf) template conveniently derivatized. Pioneer procedures for the synthesis of 2 involved the pyrolysis of D-Gal under reduced pressure [11,12] and the acid treatment under heating [13], with the subsequent tedious separation from several byproducts, including the pyranosic analogue 1. Compound 2 was thus obtained in very low yield. More recently, 2 was obtained in 32% yield by heating with a resin as an acid catalyst. Despite the greater smoothness of the method, byproducts were also formed, rendering the purification difficult [14].
Compound 2 obtained by these procedures was used to afford polymers to explore their possible applications in the field of biochemistry and pharmacology, as their properties differ from those of the corresponding monosaccharides, and they have a high density of functional groups that can be modified to obtain novel materials [14,15]. Benzylated 2 was polymerized under cationic conditions, which afforded a material not completely characterized, presumably formed by β-D-Galf units [15]. Free compound 2, as well as the D-Manf and D-Glcf analogues, was also polymerized under cationic conditions to yield a hyperbranched polysaccharide with α- and β-linked pyranosidic and furanosidic units [14].
On the other hand, when compound 2 was envisioned as a D-Galf template, the synthesis was devised starting from convenient derivatives of D-Galf in order to avoid the presence of 1. For example, compound 2 was synthesized in the past in six steps from galactose (Scheme 1) [16]. The 1,6-ring closure was produced by the O-debenzylation of the 6-hydroxy group of 4 and the nucleophilic attack of this hydroxy group to C-1, promoted by SnCl4. An optimized synthesis of 2 following this strategy has recently been described with an overall yield of 48% comprising several column chromatography purification steps [17].
Scheme 1:
Reported synthesis of 1,6-anhydro-α-D-galactofuranose [16,17].
Scheme 1:
Reported synthesis of 1,6-anhydro-α-D-galactofuranose [16,17].
The essential role of galactofuranose in the antigenic response of various pathogenic microorganisms [18-20] has triggered the interest for the development of synthetic methods for D-Galf precursors and efficient galactofuranosylation methods [21-25]. D-Galf units have been shown to be O-glycosidically linked to other D-Galf units by 1→6 linkages in many natural structures, e.g., in pathogenic Mycobateria and Aspergillius spp and others [25-28]. Benzoylated compound 5 is a good precursor of D-Galf derivatives with differentially protected hydroxy groups at position 1 and 6, for example the diacetyl derivative 6 obtained by the acetolysis of 5 (Scheme 1) [16]. In this way, compound 5 would give access to donors in which the 6-position could subsequently be manipulated for the construction of a 1→6 linkage. Based on this strategy, Ning and co-workers synthesized the β-(1→6)-linked hexasaccharide 7[29], and Kiessling‘s group developed the synthesis of compounds 8.used for the characterization of GlfT2, one of the two galactofuranosyl transferases involved in the biosynthesis of D-Galf-containing molecules (Figure 2) [30,31].
Figure 2:
Examples of glycobiological tools synthesized from compound 5[29-31].
Figure 2:
Examples of glycobiological tools synthesized from compound 5 [29-31].
Our laboratory has long been involved in the development of new galactofuranosyl derivatives and galactofuranosylation methodologies [32]. In this context, we herein report on an efficient three-step synthesis of 1,6-anhydro-α-D-galactofuranose (2) from per-O-TBS-β-D-galactofuranose (9) as a more efficient alternative to existing methods.
Results and Discussion
In the framework of our project for the development of galactofuranosyl derivatives and glycosylation methods, we have reported the synthesis of per-O-TBS-β-D-galactofuranose (9), a convenient precursor of D-Galf units, and its glycosylation via the in situ generation of galactofuranosyl iodide 10 (Scheme 2) [32-35]. Galactofuranosyl iodides were not previously described, and 10 proved to be useful for the synthesis of several D-Galf-containing molecules (Scheme 2) [32].
Scheme 2:
D-Galactofuranosylation by the glycosyl iodide method [32-35].
Scheme 2:
D-Galactofuranosylation by the glycosyl iodide method [32-35].
The reported procedure consisted in the treatment of compound 9 with 1.2 equiv of TMSI in anhydrous CH2Cl2 at 0 ºC until the total conversion of 7 into two lower moving products was observed by TLC: the 1-iodo intermediate 10 (Rf = 0.70, 10:1 hexane/EtOAc) and 2,3,5,6-tetra-O-TBS-α,β-D-galactofuranose (Rf = 0.54) formed as a result of the hydrolysis of 10 on the silica gel plate. The addition of simple alcohols or partially protected sugars as acceptors and EtN(iPr)2 as an acid scavenger led to the complete consumption of both compounds and afforded the corresponding glycosides (Scheme 2) [32]. With an excess of TMSI, a third product (Rf = 0.62) was formed, which was not consumed during the reaction and was still present in the product mixture after the work-up. The 1H NMR spectrum of this product showed a doublet at δ 5.06 with a relatively large J1,2 value (4.5 Hz). This signal correlated with a signal at 98.4 ppm in the 13C NMR spectrum, both indicative of the α-configuration. Signals corresponding to C-5 and C-6 showed similar chemical shifts. The signal corresponding to C-6 (δ 65.9) was shifted slightly downfield compared to the same signal in compound 9 (64.7 ppm), while the signal corresponding to C-5 (64.2 ppm) was significantly deshielded (10 ppm) with respect to C-5 of compound 9. No aglycone signals were observed. In order to elucidate the structure of this compound we O-desilylated it by treatment with n-Bu4NF (TBAF) in THF [36]. The product obtained (96%) was faster moving than galactose on TLC (Rf = 0.60, 7:1:2 n-PrOH/NH3/H2O) and showed 1H and 13C NMR spectra coincident with the data reported for 1,6-anhydro-α-D-galactofuranose (2) [16,37]. With the objective of optimizing the conditions for glycosylations via iodide 10, the formation of 12 was suppressed by strict control of the TMSI amount employed.
Taking into consideration how easily compound 12 was obtained and the versatility of anhydro sugars as intermediates for the preparation of biologically important oligosaccharides [3], we decided to investigate the conditions to obtain it as a main product. We reasoned that during the treatment of 9 with TMSI, in addition to the formation of the anomeric iodide, the 6-hydroxy group could be desilylated by the acid medium developed during the iodide formation (10 → 11). Then, the free 6-hydroxy group could carry out an intramolecular attack of the anomeric carbon, with iodide as a good leaving group, affording the 1,6-anhydro derivative 12 (Scheme 3).
Scheme 3:
Synthesis of 1,6-anhydro-α-D-galactofuranose from per-O-tert-butyldimethylsilyl-D-Galf.
Scheme 3:
Synthesis of 1,6-anhydro-α-D-galactofuranose from per-O-tert-butyldimethylsilyl-D-Galf.
By treatment of 9 with an excess of TMSI (2.25 equiv) in CH2Cl2 at room temperature for 5 h, compound 12 was obtained as a single product in 65% yield. Conducting the reaction at low temperature instead (−20 °C), a lower moving product was detected, presumable 11, which could not be isolated. The use of molecular sieves, which improve the reaction of 10 with alcohols or complex acceptors [32-34], should be avoided in this case as it slows down the reaction. Based on monitoring the reaction by 1H NMR (CDCl3) spectroscopy we observed that the anomeric signal of 9 (5.15 ppm, J1,2 2.6 Hz) was slowly transformed into a broad singlet at 6.53 ppm, corresponding to H-1 of 10[32], followed by the transformation into the anomeric signal of 12 (5.06 ppm, J1,2 4.5). However, the deprotection of the 6-hydroxy group of galactofuranosides affects the pattern of H-6 and H-6’ in the 1H NMR spectrum, effectively equalizing them, as was shown before [38]. During the reaction, the pair of double-doublets of H-6 and H-6’ (3.68 and 3.54 ppm) of compound 9[32] were transformed in a double-doublet (3.72 ppm) and an apparent triplet (3.60 ppm), corresponding to the H-6 and H-6’ of 12[32]. In between these two signals an intense doublet corresponding to equivalent H-6,6´ (3.58 ppm) of 11 was observed, in accordance with the behavior of other free HO-galactofuranosides [38], which supports the intermediate formation of 11.
The treatment of 9 with SnCl4 afforded compound 12, but in a lower yield due to the O-desilylation of another hydroxy group. The addition of BF3·OEt2 to recently formed 10 resulted in the formation of compound 12.
Several factors favor the formation of the bicyclic system of compound 12, such as the galactose structure itself, the presence of a good leaving group at C-1, and the electron-donating nature of the TBS groups. Thus, while compounds 13[38] and 14[39] were prepared by treatment with Lewis acids of fully protected precursors and proved to be stable and therefore useful as synthetic intermediates, attempts to prepare compound 15 by treatment with BF3·OEt2 of the persilylated precursor failed and inevitably led to the anhydro derivative 12. Moreover, whereas treatment with TFA/THF/H2O 90:5:2.5 of 4-nitrophenyl per-O-TBS-α-D-Araf afforded 16, 4-nitrophenyl per-O-TBS-β-D-galactofuranoside did not lead to compound 15 under the same conditions and compound 12 was obtained instead (Figure 3).
Figure 3:
Furanosic derivatives with free primary hydroxy group.
Figure 3:
Furanosic derivatives with free primary hydroxy group.
The O-desilylation of 12 was performed by treatment with n-Bu4F as previously optimized [32-34], affording compound 2 in almost quantitative yield (Scheme 3).
Conclusion
In conclusion, we have described a new and concise procedure for the synthesis of the 1,6-anhydro derivatives 2 and 12, the key step of which proceeds by a cascade set of three consecutive reactions. The method compares well to existing methods and by avoiding cumbersome steps, such as a benzylation and several column chromatography purifications, is an effective approach. Compounds 2 and 12 represent profitable intermediates to easily access donors and acceptors for the synthesis of Galf-containing molecules as biochemical tools.
Experimental
General methods
Analytical thin-layer chromatography (TLC) were performed on Silica Gel 60 F254 (Merck) aluminum supported plates (layer thickness 0.2 mm) with solvent systems given in the text. Visualization of the spots was effected by exposure to UV light and charring with a solution of 10% (v/v) sulfuric acid in EtOH containing 0.5% p-anisaldehyde. Column chromatography was carried out with Silica Gel 60 (230–400 mesh, Merck). Optical rotations were measured with a Perkin-Elmer 343 digital polarimeter. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker AMX 500 spectrometer. Assignments of 1H and 13C were assisted by 2D 1H COSY and HSQC experiments. High resolution mass spectra (HRMS–ESI+) were recorded in a Bruker micrOTOF-Q II spectrometer.
2,3,5-Tri-O-tert-butyldimethylsilyl-1,6-anhydro-α-D-galactofuranose (12). A solution of 9[32] (0.90 g, 1.20 mmol) in anhydrous CH2Cl2 (15 mL) was cooled to 0 ºC and stirred for 10 min under Ar. Then, iodotrimethylsilane (2.25 equiv, 0.38 mL, 2.70 mmol) was slowly added by using a syringe (10 min) while stirring was continued at 0 ºC. The reaction was allowed to reach room temperature (18–25 °C) and stirred until TLC monitoring showed the complete transformation of 9, first in two products with Rf = 0.70 and 0.54 (10:1 hexane-EtOAc), then the transformation of both products in one with Rf = 0.62 (5 h). The solution was diluted with CH2Cl2 (250 mL), washed with NaHCO3 (ss) and water, dried (Na2SO4), and concentrated. The residue was purified by column chromatography (98.6:1.4 → 2:1, hexane–EtOAc) affording compound 12 as an amorphous solid (0.272 g, 65%). The analytical data of 12 were identical with those described in ref. [32]: [α]D +42 (c 1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.05 (d, J = 4.5 Hz, 1H, H-1), 4.20 (d, J = 1.8 Hz, 1H, H-3), 4.15 (dd, J = 1.8, 4.5 Hz, 1H, H-2), 3.97 (ddd, J = 4.3, 6.3, 10.5 Hz, 1H, H-5), 3.91 (broad d, J = 4.0 Hz, 1H, H-4), 3.72 (ddd, J = 1.5, 6.2, 10.5 Hz, 1H, H-6), 3.60 (apparent t, J = 10.7 Hz, 1H, H-6′), 0.94–0.86 (SiC(CH3)3), 0.12–0.04 (Si(CH3)2); 13C NMR (125,8 MHz, CDCl3) δ 98.4 (C-1), 85.3 (C-4), 83.2 (C-2), 77.6 (C-3), 65.9 (C-6), 64.2 (C-5), 25.84, 25.89, 25.6 (SiC(CH3)3), 17.9 (SiC(CH3)3), −4.49, −4.57, −4.64, −4.68, −4.93, −5.02 (Si(CH3)2); Anal. calcd for C24H52O5Si3: C, 57.09; H, 10.38; found: C, 56.90; H, 10.52.
1,6-Anhydro-α-D-galactofuranose (2). To a solution of 12 (0.12 g, 0.22 mmol) in freshly distilled THF (7 mL), cooled at 0 °C, (n-Bu)4NF (12 equiv, 2.32 g, 8.88 mmol) was added [36]. The solution was allowed to reach room temperature and then stirring was continued for 3 h until TLC monitoring showed the complete consumption of the starting material. The solution was diluted with water (50 mL), extracted with CH2Cl2 (2 × 30 mL), and the aqueous phase was concentrated under vacuum. Purification of the residue by column chromatography (20:1 AcOEt/hexane) afforded 2 (0.036 g, 95%), [α]D +54 (c 1.0, H2O), lit. [16] [α]D +54; 1H NMR (500 MHz, D2O) δ 5.31 (d, J = 4.6 Hz, 1H, H-1), 4.26–4.23 (m, 2H, H-2, H-3), 4.19 (broad d, J = 4.2 Hz, 1H, H-4), 4.08–3.99 (m, 2H, H-5, H-6), 3.55 (apparent t, J = 10.4 Hz, 1H, H-6′); 13C NMR (125.8 MHz, D2O) δ 98.6 (C-1), 85.2 (C-4), 80.9 (C-2), 75.4 (C-3), 65.6 (C-6), 62.7 (C-5).
Supporting Information
Supporting Information File 1: 1H and 13C NMR spectra of compounds 2 and 12.
We are indebted to the University of Buenos Aires and the National Research Council of Argentina (CONICET). C.M. is research members of CONICET. L.B. was supported by a fellowship from CONICET.
Bols, M. Carbohydrate Building Blocks; Wiley: Chichester, 1996.
Return to citation in text:
[1]
Kulkarni, S. S.; Lee, J.-C.; Hung, S.-C. Curr. Org. Chem.2004,8, 475–509. doi:10.2174/1385272043485800
Return to citation in text:
[1]
[2]
[3]
Lafont, D.; Boullanger, P.; Cadas, O.; Descotes, G. Synthesis1989, 191–194. doi:10.1055/s-1989-27192
Return to citation in text:
[1]
Boons, G.-J.; Isles, S.; Setälä, P. Synlett1995, 755–756. doi:10.1055/s-1995-5057
Return to citation in text:
[1]
Skorupowa, E.; Dmochowska, B.; Madaj, J.; Kasprzykowski, F.; Sokolowski, J.; Wiśniewski, A. J. Carbohydr. Chem.1998,17, 49–59. doi:10.1080/07328309808005768
Return to citation in text:
[1]
Lauer, G.; Oberdofer, F. Angew. Chem., Int. Ed. Engl.1993,32, 272–273. doi:10.1002/anie.199302721
Return to citation in text:
[1]
Nicolaou, K. C.; Seitz, S. P.; Papahatjis, D. P. J. Am. Chem. Soc.1983,105, 2430–2434. doi:10.1021/ja00346a053
Return to citation in text:
[1]
Xue, J.; Guo, Z. Tetrahedron Lett.2001,42, 6487–6489. doi:10.1016/S0040-4039(01)01354-5
And references cited therein.
Return to citation in text:
[1]
Chun, Y.; Yan, S.; Li, X.; Ding, N.; Zhang, W.; Wang, P.; Li, M.; Li, Y. Tetrahedron Lett.2011,52, 6196–9198. doi:10.1016/j.tetlet.2011.09.055
And references therein.
Return to citation in text:
[1]
Alexander, B. H.; Dimler, R. J.; Mehltretter, C. L. J. Am. Chem. Soc.1951,73, 4658–4659. doi:10.1021/ja01154a048
Return to citation in text:
[1]
Hann, R. M.; Hudson, C. S. J. Am. Chem. Soc.1941,63, 2241–2242. doi:10.1021/ja01853a061
Return to citation in text:
[1]
Richtmyer, B. H. Methods Carbohydr. Chem.1963,2, 390–394.
Return to citation in text:
[1]
Hoai, N. T.; Sasaki, A.; Sasaki, M.; Kaga, H.; Kakuchi, T.; Satoh, T. Biomacromolecules2011,12, 1891–1899. doi:10.1021/bm2002413
Return to citation in text:
[1]
[2]
[3]
Lin, J. W.-P.; Schuerch, C. Macromolecules1972,5, 656–657. doi:10.1021/ma60029a026
Return to citation in text:
[1]
[2]
Sakar, S. K.; Choudhury, A. K.; Mukhopadhyay, B.; Roy, N. J. Carbohydr. Chem.1999,18, 1121–1130. doi:10.1080/07328309908544059
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Sakar, S. K.; Choudhury, A. K.; Hirsch, J.; Roy, N. Synthesis of 1,6-anhydro-2,3,5-tri-O-benzoyl-α-D-galactofuranose. In Carbohydrate Chemistry. Proven Synthetic methods; Kováč, P., Ed.; CRC Press: Boca Raton, USA, 2011; Vol. 1, pp 269–278.
Return to citation in text:
[1]
[2]
de Lederkremer, R. M.; Colli, W. Glycobiology1995,5, 547–552. doi:10.1093/glycob/5.6.547
Return to citation in text:
[1]
Pedersen, L. L.; Turco, S. J. Cell. Mol. Life Sci.2003,60, 259–266. doi:10.1007/s000180300021
Return to citation in text:
[1]
Marino, C.; de Lederkremer, R. M. Galactose configurations in Nature with emphasis on the biosynthesis of Galactofuranose in glycans. In Galactose: Structure and Function in Biology and Medicine; Pomin, V. H., Ed.; Nova Publisher: New York, USA, 2014; pp 107–134.
Return to citation in text:
[1]
Peltier, P.; Euzen, R.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Carbohydr. Res.2008,343, 1897–1923. doi:10.1016/j.carres.2008.02.010
Return to citation in text:
[1]
Richards, M. R.; Lowary, T. L. ChemBioChem2009,10, 1920–1938. doi:10.1002/cbic.200900208
Return to citation in text:
[1]
Akihiro, I.; Lowary, T. Trends Glycosci. Glycotechnol.2011,23, 134–152. doi:10.4052/tigg.23.134
Return to citation in text:
[1]
Marino, C.; Baldoni, L. ChemBioChem2014,15, 188–204. doi:10.1002/cbic.201300638
Return to citation in text:
[1]
Marino, C.; Gallo-Rodriguez, C.; de Lederkremer, R. M. Galactofuranosyl containing glycans: occurrence, synthesis and biochemistry. In Glycans: Biochemistry, Characterization and Applications; Mora-Montes, H. M., Ed.; Nova Science Publisher: Boca Raton, USA, 2012; pp 207–268.
Return to citation in text:
[1]
[2]
Crick, D. C.; Mahaprata, S.; Brennan, P. J. Glycobiology2001,11, 107R–118R. doi:10.1093/glycob/11.9.107R
Return to citation in text:
[1]
Besra, G. S.; Khoo, K.-H.; McNeil, M. R.; Dell, A.; Morris, H. R.; Brennan, P. J. Biochemistry1995,34, 4257–4266. doi:10.1021/bi00013a015
Return to citation in text:
[1]
Bhamidi, S.; Scherman, M. S.; Rithner, C. D.; Prenni, J. E.; Chatterjee, D.; Koo, K.-H.; McNeil, M. R. J. Biol. Chem.2008,283, 12992–13000. doi:10.1074/jbc.M800222200
Return to citation in text:
[1]
Mariño, K.; Marino, C.; de Lederkremer, R. M. Anal. Biochem.2002,301, 325–328. doi:10.1006/abio.2001.5508
Return to citation in text:
[1]
[2]
[3]
Sauvageau, J.; Foster, A. J.; Khan, A. A.; Chee, S. H.; Sims, I. M.; Timmer, M. S. M.; Stocker, B. L. ChemBioChem2012,13, 2416–2424. doi:10.1002/cbic.201200468
Return to citation in text:
[1]
Marino, C.; Gallo-Rodriguez, C.; de Lederkremer, R. M. Galactofuranosyl containing glycans: occurrence, synthesis and biochemistry. In Glycans: Biochemistry, Characterization and Applications; Mora-Montes, H. M., Ed.; Nova Science Publisher: Boca Raton, USA, 2012; pp 207–268.
Marino, C.; Gallo-Rodriguez, C.; de Lederkremer, R. M. Galactofuranosyl containing glycans: occurrence, synthesis and biochemistry. In Glycans: Biochemistry, Characterization and Applications; Mora-Montes, H. M., Ed.; Nova Science Publisher: Boca Raton, USA, 2012; pp 207–268.
Besra, G. S.; Khoo, K.-H.; McNeil, M. R.; Dell, A.; Morris, H. R.; Brennan, P. J. Biochemistry1995,34, 4257–4266. doi:10.1021/bi00013a015
28.
Bhamidi, S.; Scherman, M. S.; Rithner, C. D.; Prenni, J. E.; Chatterjee, D.; Koo, K.-H.; McNeil, M. R. J. Biol. Chem.2008,283, 12992–13000. doi:10.1074/jbc.M800222200
Marino, C.; de Lederkremer, R. M. Galactose configurations in Nature with emphasis on the biosynthesis of Galactofuranose in glycans. In Galactose: Structure and Function in Biology and Medicine; Pomin, V. H., Ed.; Nova Publisher: New York, USA, 2014; pp 107–134.
Sauvageau, J.; Foster, A. J.; Khan, A. A.; Chee, S. H.; Sims, I. M.; Timmer, M. S. M.; Stocker, B. L. ChemBioChem2012,13, 2416–2424. doi:10.1002/cbic.201200468


![[1860-5397-10-172-1]](/bjoc/content/figures/1860-5397-10-172-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-10-172-i1]](/bjoc/content/inline/1860-5397-10-172-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-10-172-2]](/bjoc/content/figures/1860-5397-10-172-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-10-172-i2]](/bjoc/content/inline/1860-5397-10-172-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-10-172-i3]](/bjoc/content/inline/1860-5397-10-172-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-10-172-3]](/bjoc/content/figures/1860-5397-10-172-3.svg?scale=2.0&max-width=1024&background=FFFFFF)