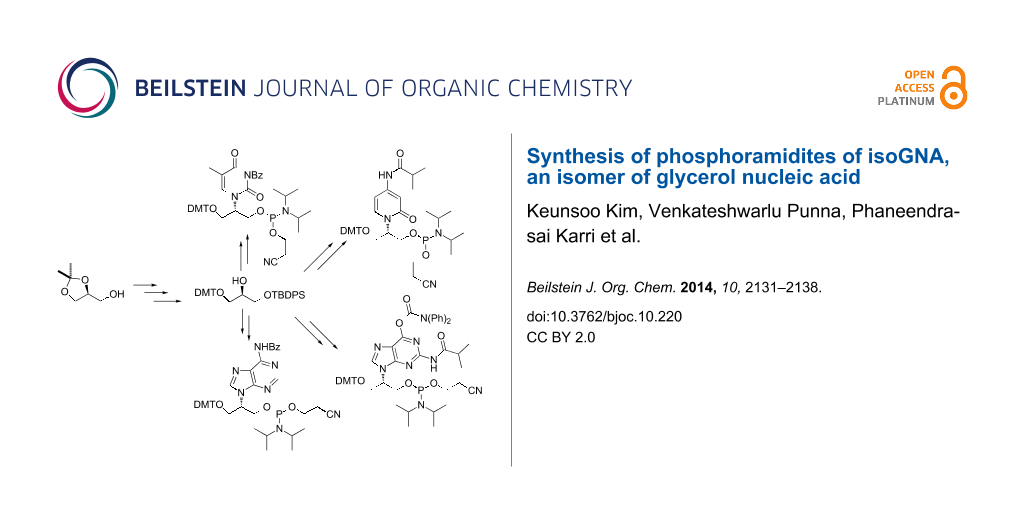
Abstract
IsoGNA, an isomer of glycerol nucleic acid GNA, is a flexible (acyclic) nucleic acid with bases directly attached to its linear backbone. IsoGNA exhibits (limited) base-pairing properties which are unique compared to other known flexible nucleic acids. Herein, we report on the details of the preparation of isoGNA phosphoramidites and an alternative route for the synthesis of the adenine derivative. The synthetic improvements described here enable an easy access to isoGNA and allows for the further exploration of this structural unit in oligonucleotide chemistry thereby spurring investigations of its usefulness and applicability.

Graphical Abstract
Introduction
Acyclic nucleic acids have garnered a lot of attention and are becoming an important component in nucleic acid chemistry [1-3]. Lately, there has been an explosion in the number of investigated candidates [4-7]. We have recently reported on the base-pairing properties of isoGNA, an isomer of glycerol derived nucleic acid (Figure 1) [8]. Our motivation was driven by the question “how structurally simple and minimal can an oligonucleotide be and still exhibit base-pairing?” When we studied isoGNA we were surprised to find that the base-pairing properties were unpredictably different from other closely related acyclic nucleic acids [8]. This indicated that the nature of the backbone exerts a strong influence on the disposition of the nucleobases, and there seems to be no straightforward correlation between the nature of the backbone and base-pairing properties [9].
![[1860-5397-10-220-1]](/bjoc/content/figures/1860-5397-10-220-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A constitutional and conformational (idealized) representation of isoGNA, an isomer of glycerol nucleic acid.
Figure 1: A constitutional and conformational (idealized) representation of isoGNA, an isomer of glycerol nuc...
We are continuing our investigation of isoGNA oligonucleotides within the context of chimeric sequences and collaborative intercalation studies and thus have a continuing need for the synthesis of isoGNA building blocks. While iso-glycerol nucleosides are known [10-15], the published synthetic routes and efficiencies of these vary widely and primarily depend on the nature of the nucleobase. Herein, we detail our synthetic efforts and the current improvements for the synthesis of all four isoGNA nucleosides and their phosphoramidites.
Results and Discussion
Initial synthetic routes
We started with commercially available (S)-solketal (1), which was protected as the benzyl ether 2. Ketal deprotection followed by the silylation of the primary hydroxy group with tert-butyldiphenylsilyl chloride provided the substrate 4, which is suitable for the introduction of the canonical nucleobases by a SN2 displacement reaction. At this point, we chose the Mitsunobu reaction (Scheme 1), which has been widely employed [16]. The reaction with the N3-benzoyl protected thymine delivered 5 in good yield. However, the reaction with N6-benzoyladenine proved problematic and no product was detected. Therefore, in the adenine case, we tried a direct SN2 reaction of N6-benzoyladenine with tosyl derivative 6, which was only moderately successful (30% yield of 7). Unexpectedly, in both the thymine and adenine cases, the subsequent debenzylation reaction failed under a variety of conditions (H2, 10% Pd/C, MeOH or EtOAc; Pd/C, DMF, cyclohexadiene; 10% Pd/C, NH4HCOO, acetone, reflux; 10% Pd-black, DMF, cyclohexadiene; (CH3)3SiI, CHCl3, 25 °C), for reasons that were not obvious to us. Consequently, we abandoned this approach and pursued a different protecting group approach.
![[1860-5397-10-220-i1]](/bjoc/content/inline/1860-5397-10-220-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Initial routes towards the synthesis of iso-glycerol nucleosides.
Scheme 1: Initial routes towards the synthesis of iso-glycerol nucleosides.
We silylated 1 to afford 8 whose ketal was deprotected followed by tritylation to give the derivative 10, which is expected to be suited for the introduction of nucleobases by an SN2 reaction (Scheme 2). Once again, the Mitsunobu reaction with thymine proceeded smoothly. However, the reaction with N6-benzoyladenine proved problematic and did not proceed even with the tosyl derivative 14. The solubility issues with benzoylated adenine and the bulk of the O-TBDPS group may be the underlying reasons behind this difficulty. Therefore, we proposed to use a doubly N6-protected adenine, the N6-benzoyl-Boc derivative 16, which was synthesized starting from N6-benzoyladenine in two steps. N6-Benzoyladenine was treated with 2.6 equiv (Boc)2O to afford N6-benzoyl-N6,N9-di-Boc-adenine 15; sat. aq NaHCO3/MeOH (1:2) conditions cleaved the N9-Boc group of 15 specifically [17,18] to yield 16. The use of 16 in the subsequent Mitsunobu reaction afforded 68% of the desired product 17. With derivatives 11 and 17 in hand we proceeded to prepare the corresponding phosphoramidite derivatives 13 and 19, respectively, under standard conditions as outlined in Scheme 2.
![[1860-5397-10-220-i2]](/bjoc/content/inline/1860-5397-10-220-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of thymine and adenine containing phosphoramidites of isoGNA.
Scheme 2: Preparation of thymine and adenine containing phosphoramidites of isoGNA.
We proceeded to prepare the cytosine- and guanine-containing phosphoramidite by using the tritylated intermediate 10 as the starting point (Scheme 3). In the cytosine series we found that the normal N4-benzoyl or N4-acetyl protected cytosine did not give satisfactory yields under the Mitsunobu conditions. The best yields were achieved with N4-isobutyrylcytosine as the nucleophile [19-21]. In the guanine series, the N2-isobutyryl, O6-diphenylcarbamoyl protected guanine worked well (Scheme 3) [22].
![[1860-5397-10-220-i3]](/bjoc/content/inline/1860-5397-10-220-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of guanine- and cytosine-containing phosphoramidites of isoGNA.
Scheme 3: Preparation of guanine- and cytosine-containing phosphoramidites of isoGNA.
With all the four phosphoramidites in hand we proceeded to the automated synthesis of the oligomers, which proceeded smoothly with ≥95% coupling efficiency as determined by trityl assays. It was at the stage of the removal of the protecting groups to afford the free oligos that we encountered a severe problem with the adenine derivative. While the aqueous ammonia/methylamine base deprotection removed all base labile protecting groups (acetate, isobutyrate, diphenylcarbamoyl groups), the N6-Boc protecting group on adenine was impervious. We tried different acidic conditions (trifluoroacetic acid, acetic acid with heat, AlCl3 [23-25]), but all of these conditions started to degrade the oligomer without completely removing the tert-Boc group (as seen by MALDI–TOF mass spectral data in combination with ion-exchange HPLC monitoring) [8].
Improved synthesis of adenine-isoGNA building block
We went back in our synthesis in order to address the adenine protecting group problem and to re-work the inefficient Mitsunobu reaction of N6-benzoyladenine. We explored various conditions (different solvents, temperature etc.) and found that the sonication [26] of the reaction mixture by using N6-benzoyladenine in anhydrous dioxane led to respectable yields (34–36%) of 26 accompanied by a (unidentified) byproduct (Scheme 4). Without sonication no product 26 was formed (by TLC).
![[1860-5397-10-220-i4]](/bjoc/content/inline/1860-5397-10-220-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of adenine containing phosphoramidite of isoGNA.
Scheme 4: Preparation of adenine containing phosphoramidite of isoGNA.
However, the yield of the sonication reaction varied capriciously and the isolation of the compound was complicated by interference from the DEAD side-products during column chromatographic purification. We did manage to get enough amounts of 26 to prepare the phosphoramidite 28, and proceeded with the oligomer synthesis for our studies. However, this was far from satisfactory, and the continued demand for isoGNA oligomers within our group and our collaborative work prompted us to look for an improvement of the preparation of the adenine building block. Considering the possible reasons for the inefficient reaction of the protected adenine derivatives in the Mitsunobu reaction, we focused on the modification of adenine in order to improve its solubility.
We considered three different derivatives of adenine based on increased solubility: 6-chloroadenine has been utilized to overcome the solubility and regioselectivity (N7 versus N9 nucleosidation) issues. Yet, the conversion of the chloro to the NH2 group is not efficient enough to consider this option an attractive one [27]. The second option was the N6,N6-dibenzoyladenine derivative, but this compound was found to be highly unstable for isolation and handling (one of the N6-benzoyl group falls off very easily). The third option was the use of the N6,N6-di-Boc-adenine derivative, even though this entails the difficulty in removing these Boc groups, which are incompatible with the oligonucleotide backbone. Therefore, we considered the option of removing these Boc groups after the Mitsunobu reaction is performed, and to introduce the N6-benzoyl group before the oligo stage.
The Schneller group has reported that N6-amino-di-Boc-protected adenine undergoes an efficient reaction under classic Mitsunobu conditions as a result of its increased solubility [28]. The regioselective attack at the N9-position is a consequence of the steric bulk of the di-Boc protection at the N6-position, which renders the N7-position inaccessible. As expected, a high yield was observed under mild conditions with substrates containing a secondary alcohol [28].
Di-Boc protected adenine was readily prepared via protection and followed by selective deprotection according to a literature procedure [17,18]. The Mitsunobu reaction between the di-Boc-adenine and 10 was investigated, and it was found that dioxane as a solvent almost doubled the yield (74%) compared to THF (36%; Scheme 5). Interestingly, sonication afforded no tangible advantage. Doubling the amount of DIAD or di-Boc-adenine or PPh3 did not give increased yields. The desired product 29 was easily purified on silica gel due to its lower polarity (when compared with the N6-benzoyladenine reaction).
![[1860-5397-10-220-i5]](/bjoc/content/inline/1860-5397-10-220-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mitsunobu reaction with the di-Boc-adenine.
Scheme 5: Mitsunobu reaction with the di-Boc-adenine.
Initially, we tried to simultaneously deprotect both Boc groups and the TBDPS group in the presence of TBAF in a one-pot reaction [29]. However, only the TBDPS group was removed and a quantitative amount of mono-protected compound 30 was produced (Scheme 6). The fluoride ion was not effective enough to remove at least one of the Boc groups in 29. We checked the deprotection conditions for the removal of the Boc moiety by fluoride ions on the model substrate N6,N6-di-Boc-adenine, and found that it was very challenging because free adenine was not afforded even under reflux with 1 M TBAF for 4 days.
![[1860-5397-10-220-i6]](/bjoc/content/inline/1860-5397-10-220-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: An attempt to remove both Boc and silyl groups simultaneously.
Scheme 6: An attempt to remove both Boc and silyl groups simultaneously.
Finally, we decided to remove the Boc groups on adenine with trifluoroacetic acid (TFA), even though we also expected the loss of the DMTr group on 29. When 29 was treated with 20% of TFA in CH2Cl2, a mixture of 32 and 33 formed after 4 h. The TBDPS moiety was found to be stable under these conditions (Table 1, entry 1). 50% of TFA in CH2Cl2 completely removed both Boc groups on adenine at either 4 °C or rt to afford the desired 33 in 87% yield.
Table 1: Result of Boc deprotection of 29 with TFA.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-220-i8.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Ratio of TFA (%) | Temp.a | Time (h) |
Ratio (%)b
(31:32:33) |
Yield | |
|---|---|---|---|---|---|---|
| 1 | 20 | 0 °C to rt | 4 | 0:33:66 | – | |
| 2 | 50 | 0 °C to 4 °C | 2 | 0:20:80 | – | |
| 4 | 0:0:100 | – | ||||
| 3c | 50 | 0 °C to rt | 2 | 0:0:100 | 87% | |
aTFA (99%) was added under ice-bath (−2 to 2 °C) cooling. bThe ratio is roughly calculated by the intensity of visualized spots on TLC by UV lamp. cThis reaction was performed on a large scale.
The trityl group was reintroduced on 33 under standard conditions to give 34, and subsequent benzoylation afforded 26 in overall good yields (Scheme 7). The NMR spectra of 26 were in good agreement with those data obtained previously [8] via Mitsunobu reaction with N6-benzoyladenine.
![[1860-5397-10-220-i7]](/bjoc/content/inline/1860-5397-10-220-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The synthesis of 26 via tritylation and benzoylation.
Scheme 7: The synthesis of 26 via tritylation and benzoylation.
Overall, 26 was afforded in 43% yield from the Mitsunobu reaction of 10 with N6,N6-di-Boc-adenine. In addition to the slight improvement compared to the direct Mitsunobu reaction of 10 with N6-benzoyladenine, this protocol allows for the synthesis of large amounts of 26 with an exact control of the regioselectivity at the N9-position of adenine.
Having solved the “adenine problem”, we focused on each of the remaining steps and optimized the synthesis of other isoGNA phosphoramidites. For example, the deprotection of 8 to 9 was initially achieved by the treatment with conc. HCl. However, in scaling up the reaction, the loss of the silyl group was found to occur. Therefore, we sought an alternative route and found that the deprotection mediated by vanadium trichloride gave the desired product 9 cleanly in large-scale reactions [30]. The desilylation of intermediates 11, 20 and 26 was effected by the treatment of 1 M TBAF in THF at 0 °C to room temperature. In the case of guanine derivative 23, we observed that the use of the triethylamine·HF complex instead of TBAF was preferable at room temperature. The free alcohols 12, 18, 21 and 24 were phosphitylated under standard conditions to obtain the phosphoramidites suitable for the automated solid phase synthesis.
Conclusion
We have described herein improvements to and the optimization of the synthesis of isoGNA phosphoramidite building blocks. We observed that the Boc protecting group on adenine while not being compatible with oligonucleotide chemistry, can nevertheless be used as a temporary protecting group, which is extremely useful for increasing the solubility and regioselectivity of adenine in the Mitsunobu reaction. The optimized procedures described here allowed us to access a stable supply of isoGNA phosphoramidites for further investigation in our laboratory.
References
-
Vandendriessche, F.; Augustyns, K.; Van Aerschot, A.; Busson, R.; Hoogmartens, J.; Herdewijn, P. Tetrahedron 1993, 49, 7223–7238. doi:10.1016/S0040-4020(01)87200-8
Return to citation in text: [1] -
Nielsen, P.; Dreiøe, L. H.; Wengel, J. Bioorg. Med. Chem. 1995, 3, 19–28. doi:10.1016/0968-0896(94)00143-Q
Return to citation in text: [1] -
Zhang, S.; Switzer, C.; Chaput, J. C. Chem. Biodiversity 2010, 7, 245–258. doi:10.1002/cbdv.200900281
Return to citation in text: [1] -
Kumar, V.; Gore, K. R.; Pradeepkumar, P. I.; Kesavan, V. Org. Biomol. Chem. 2013, 11, 5853–5865. doi:10.1039/c3ob41244j
Return to citation in text: [1] -
Murayama, K.; Tanaka, Y.; Toda, T.; Kashida, H.; Asanuma, H. Chem. – Eur. J. 2013, 19, 14151–14158. doi:10.1002/chem.201301578
Return to citation in text: [1] -
Perlíková, P.; Karlsen, K. K.; Pedersen, E. B.; Wengel, J. ChemBioChem 2014, 15, 146–156. doi:10.1002/cbic.201300567
Return to citation in text: [1] -
Li, P.; Sun, J.; Su, M.; Yang, X.; Tang, X. Org. Biomol. Chem. 2014, 12, 2263–2272. doi:10.1039/c3ob42291g
Return to citation in text: [1] -
Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem., Int. Ed. 2013, 52, 5840–5844. doi:10.1002/anie.201300795
Return to citation in text: [1] [2] [3] [4] -
Krishnamurthy, R. Synlett 2014, 25, 1511–1517. doi:10.1055/s-0033-1340183
Return to citation in text: [1] -
Mikhailov, S. N.; Smrt, J. Collect. Czech. Chem. Commun. 1975, 40, 3080–3085. doi:10.1135/cccc19753080
Return to citation in text: [1] -
Hiller, S. A.; Zarin, D. E.; Zhuk, R. A. Nucleic Acids Res. 1976, 3, 721–728. doi:10.1093/nar/3.3.721
Return to citation in text: [1] -
Capaldi, D. C.; Eleuteri, A.; Chen, Q.; Schinazi, R. F. Nucleosides Nucleotides 1997, 16, 403–416. doi:10.1080/07328319708001358
Return to citation in text: [1] -
Cadet, G.; Chan, C.-S.; Daniel, R. Y.; Davis, C. P.; Guiadeen, D.; Rodriguez, G.; Thomas, T.; Walcott, S.; Scheiner, P. J. Org. Chem. 1998, 63, 4574–4580. doi:10.1021/jo9715231
Return to citation in text: [1] -
Rosenberg, I.; Holý, A.; Masojídoková, M. Collect. Czech. Chem. Commun. 1988, 53, 2753–2777. doi:10.1135/cccc19882753
Return to citation in text: [1] -
Vrbovská, S.; Holý, A.; Pohl, R.; Masojídoková, M. Collect. Czech. Chem. Commun. 2006, 71, 543–566. doi:10.1135/cccc20060543
Return to citation in text: [1] -
Lowe, G.; Vilaivan, T. J. Chem. Soc., Perkin Trans. 1 1997, 539–546. doi:10.1039/a603696a
Return to citation in text: [1] -
Dey, S.; Garner, P. J. Org. Chem. 2000, 65, 7697–7699. doi:10.1021/jo000983i
Return to citation in text: [1] [2] -
Porcheddu, A.; Giacomelli, G.; Píredda, I.; Carta, M.; Nieddu, G. Eur. J. Org. Chem. 2008, 5786–5797. doi:10.1002/ejoc.200800891
Return to citation in text: [1] [2] -
Antle, V. D.; Caperelli, C. A. Nucleosides Nucleotides 1999, 18, 1911–1928. doi:10.1080/07328319908044853
Return to citation in text: [1] -
Choo, H.; Chong, Y.; Chu, C. K. Org. Lett. 2001, 3, 1471–1473. doi:10.1021/ol015783n
Return to citation in text: [1] -
Shatila, R. S.; Bouhadir, K. H. Tetrahedron Lett. 2006, 47, 1767–1770. doi:10.1016/j.tetlet.2006.01.035
Return to citation in text: [1] -
Lu, W.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Org. Chem. 2007, 72, 5012–5015. doi:10.1021/jo070515+
Return to citation in text: [1] -
James, K. D.; Ellington, A. D. Tetrahedron Lett. 1998, 39, 175–178. doi:10.1016/S0040-4039(97)10509-3
Return to citation in text: [1] -
James, K. D.; von Kiedrowski, G.; Ellington, A. D. Nucleosides Nucleotides 1997, 16, 1821–1836. doi:10.1080/07328319708002536
Return to citation in text: [1] -
Pothukanuri, S.; Pianowski, Z.; Winssinger, N. Eur. J. Org. Chem. 2008, 3141–3148. doi:10.1002/ejoc.200800141
Return to citation in text: [1] -
Lepore, S. D.; He, Y. J. Org. Chem. 2003, 68, 8261–8263. doi:10.1021/jo0345751
Return to citation in text: [1] -
Perez-Perez, M.-J.; Rozenski, J.; Busson, R.; Herdewijn, P. J. Org. Chem. 1995, 60, 1531–1537. doi:10.1021/jo00111a010
Return to citation in text: [1] -
Yin, X.-q.; Li, W.-k.; Schneller, S. W. Tetrahedron Lett. 2006, 47, 9187–9189. doi:10.1016/j.tetlet.2006.10.126
Return to citation in text: [1] [2] -
Jacquemard, U.; Bénéteau, V.; Lefoix, M.; Routier, S.; Mérour, J.-Y.; Coudert, G. Tetrahedron 2004, 60, 10039–10047. doi:10.1016/j.tet.2004.07.071
Return to citation in text: [1] -
Sabitha, G.; Reddy, G. S. K. K.; Reddy, K. B.; Reddy, N. M.; Yadav, J. S. J. Mol. Catal. A: Chem. 2005, 238, 229–232. doi:10.1016/j.molcata.2005.05.028
Return to citation in text: [1]
| 8. | Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem., Int. Ed. 2013, 52, 5840–5844. doi:10.1002/anie.201300795 |
| 17. | Dey, S.; Garner, P. J. Org. Chem. 2000, 65, 7697–7699. doi:10.1021/jo000983i |
| 18. | Porcheddu, A.; Giacomelli, G.; Píredda, I.; Carta, M.; Nieddu, G. Eur. J. Org. Chem. 2008, 5786–5797. doi:10.1002/ejoc.200800891 |
| 29. | Jacquemard, U.; Bénéteau, V.; Lefoix, M.; Routier, S.; Mérour, J.-Y.; Coudert, G. Tetrahedron 2004, 60, 10039–10047. doi:10.1016/j.tet.2004.07.071 |
| 1. | Vandendriessche, F.; Augustyns, K.; Van Aerschot, A.; Busson, R.; Hoogmartens, J.; Herdewijn, P. Tetrahedron 1993, 49, 7223–7238. doi:10.1016/S0040-4020(01)87200-8 |
| 2. | Nielsen, P.; Dreiøe, L. H.; Wengel, J. Bioorg. Med. Chem. 1995, 3, 19–28. doi:10.1016/0968-0896(94)00143-Q |
| 3. | Zhang, S.; Switzer, C.; Chaput, J. C. Chem. Biodiversity 2010, 7, 245–258. doi:10.1002/cbdv.200900281 |
| 28. | Yin, X.-q.; Li, W.-k.; Schneller, S. W. Tetrahedron Lett. 2006, 47, 9187–9189. doi:10.1016/j.tetlet.2006.10.126 |
| 8. | Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem., Int. Ed. 2013, 52, 5840–5844. doi:10.1002/anie.201300795 |
| 28. | Yin, X.-q.; Li, W.-k.; Schneller, S. W. Tetrahedron Lett. 2006, 47, 9187–9189. doi:10.1016/j.tetlet.2006.10.126 |
| 8. | Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem., Int. Ed. 2013, 52, 5840–5844. doi:10.1002/anie.201300795 |
| 26. | Lepore, S. D.; He, Y. J. Org. Chem. 2003, 68, 8261–8263. doi:10.1021/jo0345751 |
| 4. | Kumar, V.; Gore, K. R.; Pradeepkumar, P. I.; Kesavan, V. Org. Biomol. Chem. 2013, 11, 5853–5865. doi:10.1039/c3ob41244j |
| 5. | Murayama, K.; Tanaka, Y.; Toda, T.; Kashida, H.; Asanuma, H. Chem. – Eur. J. 2013, 19, 14151–14158. doi:10.1002/chem.201301578 |
| 6. | Perlíková, P.; Karlsen, K. K.; Pedersen, E. B.; Wengel, J. ChemBioChem 2014, 15, 146–156. doi:10.1002/cbic.201300567 |
| 7. | Li, P.; Sun, J.; Su, M.; Yang, X.; Tang, X. Org. Biomol. Chem. 2014, 12, 2263–2272. doi:10.1039/c3ob42291g |
| 27. | Perez-Perez, M.-J.; Rozenski, J.; Busson, R.; Herdewijn, P. J. Org. Chem. 1995, 60, 1531–1537. doi:10.1021/jo00111a010 |
| 19. | Antle, V. D.; Caperelli, C. A. Nucleosides Nucleotides 1999, 18, 1911–1928. doi:10.1080/07328319908044853 |
| 20. | Choo, H.; Chong, Y.; Chu, C. K. Org. Lett. 2001, 3, 1471–1473. doi:10.1021/ol015783n |
| 21. | Shatila, R. S.; Bouhadir, K. H. Tetrahedron Lett. 2006, 47, 1767–1770. doi:10.1016/j.tetlet.2006.01.035 |
| 23. | James, K. D.; Ellington, A. D. Tetrahedron Lett. 1998, 39, 175–178. doi:10.1016/S0040-4039(97)10509-3 |
| 24. | James, K. D.; von Kiedrowski, G.; Ellington, A. D. Nucleosides Nucleotides 1997, 16, 1821–1836. doi:10.1080/07328319708002536 |
| 25. | Pothukanuri, S.; Pianowski, Z.; Winssinger, N. Eur. J. Org. Chem. 2008, 3141–3148. doi:10.1002/ejoc.200800141 |
| 17. | Dey, S.; Garner, P. J. Org. Chem. 2000, 65, 7697–7699. doi:10.1021/jo000983i |
| 18. | Porcheddu, A.; Giacomelli, G.; Píredda, I.; Carta, M.; Nieddu, G. Eur. J. Org. Chem. 2008, 5786–5797. doi:10.1002/ejoc.200800891 |
| 8. | Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem., Int. Ed. 2013, 52, 5840–5844. doi:10.1002/anie.201300795 |
| 16. | Lowe, G.; Vilaivan, T. J. Chem. Soc., Perkin Trans. 1 1997, 539–546. doi:10.1039/a603696a |
| 30. | Sabitha, G.; Reddy, G. S. K. K.; Reddy, K. B.; Reddy, N. M.; Yadav, J. S. J. Mol. Catal. A: Chem. 2005, 238, 229–232. doi:10.1016/j.molcata.2005.05.028 |
| 10. | Mikhailov, S. N.; Smrt, J. Collect. Czech. Chem. Commun. 1975, 40, 3080–3085. doi:10.1135/cccc19753080 |
| 11. | Hiller, S. A.; Zarin, D. E.; Zhuk, R. A. Nucleic Acids Res. 1976, 3, 721–728. doi:10.1093/nar/3.3.721 |
| 12. | Capaldi, D. C.; Eleuteri, A.; Chen, Q.; Schinazi, R. F. Nucleosides Nucleotides 1997, 16, 403–416. doi:10.1080/07328319708001358 |
| 13. | Cadet, G.; Chan, C.-S.; Daniel, R. Y.; Davis, C. P.; Guiadeen, D.; Rodriguez, G.; Thomas, T.; Walcott, S.; Scheiner, P. J. Org. Chem. 1998, 63, 4574–4580. doi:10.1021/jo9715231 |
| 14. | Rosenberg, I.; Holý, A.; Masojídoková, M. Collect. Czech. Chem. Commun. 1988, 53, 2753–2777. doi:10.1135/cccc19882753 |
| 15. | Vrbovská, S.; Holý, A.; Pohl, R.; Masojídoková, M. Collect. Czech. Chem. Commun. 2006, 71, 543–566. doi:10.1135/cccc20060543 |
| 22. | Lu, W.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Org. Chem. 2007, 72, 5012–5015. doi:10.1021/jo070515+ |
© 2014 Kim et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)