Abstract
The one-pot three-component reactions of 1-substituted benzimidazoles with ethyl bromoacetate and electron-deficient alkynes, in 1,2-epoxybutane, gave a variety of pyrrolo[1,2-a]quinoxalin-4-ones and pyrrolo[1,2-a]benzimidazoles. The influence of experimental conditions on the course of reaction was investigated. A novel synthetic pathway starting from benzimidazoles unsubstituted at the five membered ring, alkyl bromoacetates and non-symmetrical electron-deficient alkynes in the molar ratio of 1:2:1, in 1,2-epoxybutane at reflux temperature, led directly to pyrrolo[1,2-a]quinoxalin-4-ones in fair yield by an one-pot three-component reaction.

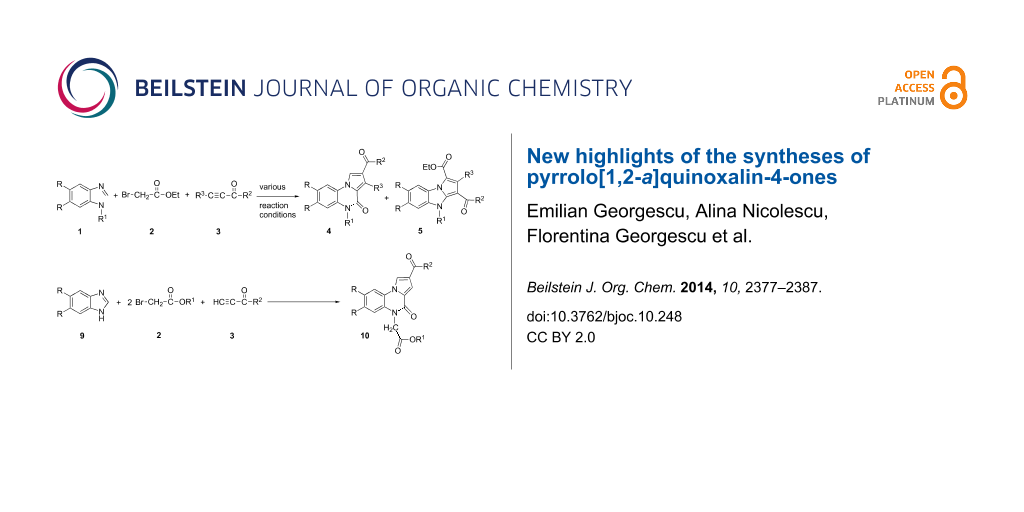
Graphical Abstract
Introduction
The pyrrolo[1,2-a]quinoxaline system has significant biological activities and is a subject fo constant interest. This skeleton is a constituent of several bioactive heterocyclic compounds that demonstrate interesting activity against Mycobacterium tuberculosis [1], anti-HIV [2], anticancer [3], and it modulates the estrogen receptor activity [4].
Synthetic methods towards pyrrolo[1,2-a]quinoxaline derivatives based on pyrroles [5], or quinoxalines [6] have been recently reviewed. Among other synthetic routes, the 1,3-dipolar cycloaddition of heterocyclic N-ylides with various activated alkynes or alkenes is an important method for constructing fused heterocyclic systems such as pyrrolo[1,2-a]quinoxaline and pyrrolo[1,2-a]benzimidazole [7-13]. The development of more efficient synthetic methods towards these compounds is an active research area [14-16].
Recently, we reported on the formation of pyrrolo[1,2-a]benzimidazoles along with pyrrolo[1,2-a]quinoxalines in the one-pot three-component reaction of 1-benzylbenzimidazoles, phenacyl bromides and non-symmetrical activated alkynes in presence of propenoxide or 1,2-epoxybutane used as acid scavenger and reaction solvent [16]. These results prompted us to further investigate 1,3-cycloaddition reactions of 1-substituted 3-(alkoxycarbonylmethyl)benzimidazolium ylides with various dipolarophiles under the same reaction conditions, aiming to explore the generality of the reaction.
The previously reported data on 1,3-cycloaddition reactions of 1-substituted 3-(alkoxycarbonylmethyl)benzimidazolium ylides with various dipolarophiles are rather contradictory. Thus, 1-alkyl-3-(methoxycarbonylmethyl)benzimidazolium bromides with dimethyl acetylenedicarboxylate (DMAD) in presence of K2CO3 in DMF [7] or in presence of triethylamine in acetonitrile [8] give a mixture of pyrrolo[1,2-a]benzimidazole (2–7%) and a compound whose formation involves the loss of an alcohol molecule for which different structures have been proposed [7,8]. The correct structure of 2,3-dicarbomethoxy-5-methylpyrrolo[1,2-a]quinoxalin-4-one and the reaction mechanism was proposed by Meth-Cohn [9].
The reactions of 1-substituted 3-(ethoxycarbonylmethyl)benzimidazolium bromides with fluoroalkenes [10] or fluorovinyl tosylates [11] in presence of K2CO3 and triethylamine in DMF at 70 °C, or with activated alkenes, such as acrylates, acrylonitrile or diethyl malonate, in presence of triethylamine and an oxidant in DMF at 90 °C, led only to the normal cycloaddition products, i.e., pyrrolo[1,2-a]benzimidazoles [12]. When the same reactions were performed with polarized alkenes, such as 2-ethoxy acrylonitrile or 1,1-bis(methylthio)-2-nitroethylene, in presence of K2CO3 in chloroform at room temperature, only pyrrolo[1,2-a]quinoxalin-4-ones resulted in fair yields [13].
Our interest in developing simple synthetic pathways towards N-bridged heterocyclic compounds [17-20] prompted us to investigate the one-pot three-component reactions of various substituted benzimidazoles with alkyl bromoacetates and electron-deficient alkynes in presence of an epoxide. Herein, we report a simple one-pot three-component synthetic procedure towards pyrrolo[1,2-a]quinoxalin-4-ones and pyrrolo[1,2-a]benzimidazoles and we describe the influence of reaction conditions on the ratio of the two final reaction products. We developed also a selective one-pot three-component synthetic pathway towards pyrrolo[1,2-a]quinoxalin-4-one derivatives starting from benzimidazole derivatives unsubstituted at the five membered ring, alkyl bromoacetates and non-symmetrical electron-deficient alkynes in the molar ratio of 1:2:1, in 1,2-epoxybutane at reflux temperature.
Results and Discussion
The one-pot three-component reaction of 1-substituted benzimidazoles 1a–c, ethyl bromoacetate 2 and non-symmetrical activated alkynes 3a–c, in almost equimolar amounts, performed in presence of 1,2-epoxybutane gave pyrrolo[1,2-a]quinoxaline-4-ones 4a–g as major reaction products. Pyrrolo[1,2-a]benzimidazoles 5b,e were isolated along with pyrrolo[1,2-a]quinoxaline-4-ones 4b,e only in some cases (Scheme 1, Table 1). All reactions have been performed by mixing the starting components at room temperature in 1,2-epoxybutane and heating the reaction mixture for 24 hours at reflux temperature. Pyrrolo[1,2-a]quinoxalin-4-one derivatives 4 were isolated from the reaction mixture by crystallization. To separate pyrrolo[1,2-a]benzimidazole derivatives 5, each filtrate was concentrated under vacuum and chromatographed on a SiO2 packed column.
The HPLC analysis of crude reaction products indicated that small amounts of pyrrolo[1,2-a]benzimidazoles 5 were formed in all reactions, but they could not be always isolated from the reaction mixtures.
Due to the high reactivity of dimethyl acetylenedicarboxylate which can react also with the starting 1-substituted benzimidazole, the one-pot three-component synthetic procedure starting from almost equimolar amounts of 1-substituted benzimidazole 1, ethyl bromoacetate and dimethyl acetylenedicarboxylate (3d) in 1,2-epoxybutane led to a complex mixture of reaction products. However, by direct reaction of 1-benzyl-3-ethoxycarbonylmethylbenzimidazolium bromide, obtained previously from 1-benzylbenzimidazole (1a) and ethyl bromoacetate (2), with dimethyl acetylenedicarboxylate (3d), in 1,2-epoxybutane at reflux temperature, the pyrrolo[1,2-a]quinoxalin-4-one (4h) was obtained as major reaction product along with a small amount of pyrrolo[1,2-a]benzimidazole (5h). Starting from 1-benzyl-5,6-dimethyl-3-ethoxycarbonylmethylbenzimidazolium bromide and dimethyl acetylenedicarboxylate 3d, in the same conditions, only pyrrolo[1,2-a]quinoxalin-4-one 4i was isolated from the reaction mixture (Scheme 1).
![[1860-5397-10-248-i1]](/bjoc/content/inline/1860-5397-10-248-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]benzimidazoles 5.
Scheme 1: Synthesis of pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]benzimidazoles 5.
The yields and melting points of newly synthesized pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]bezimidazoles 5 are presented in Table 1.
Table 1: Synthesized pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]bezimidazoles 5.
| Entry | Reaction products | |||||
|---|---|---|---|---|---|---|
| 4 | mp (°C) | Yield (%)a | 5 | mp (°C) | Yield (%)a | |
| 1 | 4a | 225–227 | 39 | – | – | – |
| 2 | 4b | 178–180 | 42 | 5b | 130–132 | 13 |
| 3 | 4c | 220–222 | 57 | – | – | – |
| 4 | 4d | 274–275 | 43 | – | – | – |
| 5 | 4e | 215–217 | 38 | 5e | 191–193 | 21 |
| 6 | 4f | 283–285 | 48 | – | – | – |
| 7 | 4g | 191–193 | 39 | – | – | – |
| 8 | 4h |
259–261
258–259 [8] |
42
12 [8] |
5h | 177–178 | 16 |
| 9 | 4i | 275–276 | 19 | – | – | – |
aYields for isolated and purified compounds.
The reaction pathway (Scheme 2) involves the quaternization of 1-substituted benzimidazoles 1 with ethyl bromoacetate (2) leading to corresponding benzimidazolium bromides 6. The attack of the bromine ion from the benzimidazolium bromide on the oxirane ring in 1,2-epoxybutane results in ring opening and generation of the benzimidazolium N-ylide 7 by the action of the alkoxide. The benzimidazolium N-ylide 7 reacts with the activated alkynes 3 to give the corresponding primary cycloadduct dihydropyrrolo[1,2-a]benzimidazoles 8. The formation of pyrrolo[1,2-a]quinoxalin-4-ones 4 involves the imidazole ring-opening, initiated by the deprotonation at C-1 of the primary cycloadducts 8, followed by ring-closure involving the carbethoxy C=O group, a previously proposed rationale [9]. The formation of pyrrolo[1,2-a]benzimidazoles 5 involves the spontaneous in situ dehydrogenation of the primary cycloadducts 8.
![[1860-5397-10-248-i2]](/bjoc/content/inline/1860-5397-10-248-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction pathway leading to the formation of pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]bezimidazoles 5.
Scheme 2: Reaction pathway leading to the formation of pyrrolo[1,2-a]quinoxalin-4-ones 4 and pyrrolo[1,2-a]be...
In order to explain the above mentioned results, we investigated the influence of reaction conditions on the ratio of the final reaction products 4 and 5 in 1,3-dipolar cycloaddition reactions of the 1-benzyl-3-(ethoxycarbonylmethyl)benzimidazolium bromide 6 (R = H, R1 = benzyl) with ethyl propiolate (3b) and DMAD (3d), in different reaction conditions reported in literature (Table 2). In these experiments, all crude reaction products were treated with aqueous solution of 5% HCl and extracted with CHCl3. The chloroformic extracts were dried, concentrated under vacuum, analyzed by HPLC and the peak areas of the final reaction products 4 to 5 were determined (Table 2).
Table 2: The influence of the reaction conditions on the final reaction products.
| Entry | Reaction conditions | Ratio of peak areasa | |
|---|---|---|---|
| 4b:5b | 4h:5h | ||
| 1 | 1,2-epoxybutane, 24 h at reflux temperature (≈62 °C) | 7.6 | 6.2 |
| 2 | NEt3 and TPCD in DMF, 4 h at 90 °Cb | 7.7 | 2.7 |
| 3 | NEt3 in acetonitrile, 4 h at reflux temperature (≈80 °C)c | 46 | 54 |
| 4 | K2CO3 in DMF, 48 h at rtd | 27 | – |
| 5 | K2CO3 + NEt3 in DMF, 24 h at 70 °Ce | 91 | – |
aCalculated from HPLC chromatograms; breaction conditions according to [12]; caccording to [8]; daccording to [7]; eaccording to [10,11].
The results suggest that in the presence of an organic and/or inorganic base the formation of pyrrolo[1,2-a]quinoxalin-4-one derivatives 4 is favored, while in a neutral medium or in the presence of oxidants, such as TPCD [12], significant quantities of pyrrolo[1,2-a]benzimidazoles 5, the normal 1,3-dipoar cycladdition product, are also formed. In this way, the low yields of pyrrolo[1,2-a]benzimidazoles 5 reported in literature [7,8] can be explained.
An easy access to pyrrolo[1,2-a]quinoxalin-4-ones 10 was provided by the one-pot three-components reaction of benzimidazoles unsubstituted at the imidazole ring 9a,b with alkyl bromoacetates 2a,b and non-symmetrical, electron-deficient alkynes 3a,b, in the molar ratio 1:2:1, in 1,2-epoxybutane at reflux temperature. This novel synthetic procedure lead directly to pyrrolo[1,2-a]quinoxalin-4-ones 10a–f, as solely reaction product, in fair yields (Scheme 3).
![[1860-5397-10-248-i3]](/bjoc/content/inline/1860-5397-10-248-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Novel synthetic pathway towards pyrrolo[1,2-a]quinoxalin-4-ones 10.
Scheme 3: Novel synthetic pathway towards pyrrolo[1,2-a]quinoxalin-4-ones 10.
A range of benzimidazole, unsubstituted at the imidazole ring and bearing various substituents on the benzoanelated ring, could be used as starting compounds. The reaction could be extended for a broad range of alkyl bromoacetates and non-symmetrical electron-deficient alkynes. Mild reaction conditions are involved, implying mixing the benzimidazole derivative with an alkyl bromoacetate and a non-symmetrical activated alkyne in the molar ratio of 1:2:1 at room temperature in 1,2-epoxybutane, then heating the reaction mixture at reflux temperature for 30 hours. All final pyrrolo[1,2-a]quinoxalin-4-one compounds have been isolated by simple, non-chromatographic methods.
The reaction pathway involves the intermediate N-alkylation of the imidazole ring with one equivalent of alkyl bromoacetate yielding 1-ethoxycarbonylmethylbenzimidazole, followed by its quaternization with the second equivalent of alkyl bromoacetate leading to 1,3-di(ethoxycarbonylmethyl)benzimidazolium bromide. The final pyrrolo[1,2-a]quinoxalin-4-ones are obtained according to the mechanism presented in Scheme 2.
The structures of newly synthesized pyrrolo[1,2-a]quinoxalin-4-ones 4 and 10, and pyrrolo[1,2-a]benzimidazoles 5 were assigned by elemental analysis, IR and NMR spectroscopy. The 1H, 13C and 15N NMR chemical shifts have been unambiguously assigned based on the following 2D NMR experiments: H,H-COSY, H,C-HSQC, H,C-HMBC, H,N-HMBC, H,H-NOESY.
In the 1H NMR spectra of pyrrolo[1,2-a]quinoxalines and pyrrolo[1,2-a]benzimidazoles the protons from the phenyl ring and the annelated benzo ring are overlapping in the region of 7–8 ppm. Based on a less used undecoupled H,C-HSQC type of spectrum we assigned for the first time the individual aromatic signals, the multiplicity and the order of magnitude of the coupling constants for these classes of compounds. The full assignments are listed in the experimental section and an example is shown in Figure 1 for compound 5h. Thus, in Figure 1, one can clearly see separated cross peaks around each 13C satellite corresponding to all 1H signals in the region of 7.0–7.6 ppm. The low intensity 13C satellites in the 1H NMR spectrum are located outside (low and high field) of the region of the main 1H signal. When extracting 1D rows from the 2D H,C-undecoupled-HSQC spectrum corresponding to each 13C signal, one can see traces showing individual 1H signals (Figure 2). The pseudo 1D spectra from Figure 2 are traces at each 13C signal around the low field 13C satellite in the 1H dimension. In contrast with the normal 1H NMR spectrum (Figure 2, bottom) the pseudo 1D 1H spectra show individual signals allowing for the determination of chemical shifts, multiplicities, and coupling constants.
![[1860-5397-10-248-1]](/bjoc/content/figures/1860-5397-10-248-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Undecoulpled H,C-HSQC spectrum for compound 5h.
Figure 1: Undecoulpled H,C-HSQC spectrum for compound 5h.
![[1860-5397-10-248-2]](/bjoc/content/figures/1860-5397-10-248-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Individual 1H signal assignments based on 13C traces from H,C-undecoulpled-HSQC spectrum around the low field 13C satellite, in comparison with the 1H NMR spectrum (bottom) for compound 5h.
Figure 2: Individual 1H signal assignments based on 13C traces from H,C-undecoulpled-HSQC spectrum around the...
For compounds 4h, 4i, 5b and 5e, the carbomethoxy respectively carbethoxy residues were assigned based on their NOE response. Thus, for compounds 4h,i the methyl protons from carbomethoxy groups situated in positions 2 and 3 were differentiated based on their NOE cross peak with the proton in position 1. For compounds 5b,e the protons from carbethoxy groups situated in positions 1 and 3 were assigned based on their NOE cross peaks with the proton in position 8, an example for 5b is shown in Figure 3.
![[1860-5397-10-248-3]](/bjoc/content/figures/1860-5397-10-248-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: NOE response as cross peaks between carbethoxy group protons and protons from positions 2 and 8 of the heterocycle in a detail from the H,H-NOESY spectrum for compound 5b.
Figure 3: NOE response as cross peaks between carbethoxy group protons and protons from positions 2 and 8 of ...
Based on the NOE assignments of various ethyl groups, we suppose that the preferred conformation in solution for the carbethoxy group in position 1 in compounds 5b,e,h is oriented towards the benzo-annelated nucleus, thus the aromatic ring current inducing a deshielding effect on the CH2 and CH3 groups. On the contrary, in compounds 10a–d we assume a solution preferred orientation of the carbethoxy group in position 5-N-CH2- of the heterocycle away from the benzo-annelated nucleus and on the same side with the carbonyl group, the latter inducing a shielding effect on the CH3 group.
Conclusion
We have demonstrated that 1,3-dipolar cycloaddition reactions of 1-benzyl-3-(alkoxycarbonylmethyl)benzimidazolium ylides with activated alkynes led to a mixture of pyrrolo[1,2-a]quinoxalin-4-ones and pyrrolo[1,2-a]benzimidazoles. Pyrrolo[1,2-a]quinoxalin-4-ones are always the major reaction product and the ratio of pyrrolo[1,2-a]quinoxalin-4-one to pyrrolo[1,2-a]benzimidazole depends on reaction conditions and reactant structures.
A selective one-pot three-component synthetic protocol providing easy access to a wide range of pyrrolo[1,2-a]quinoxalin-4-one derivatives starts from benzimidazole unsubstituted at the imidazole ring, alkyl bromoacetates and non-symmetrical electron-deficient alkynes in the molar ratio 1:2:1, in 1,2-epoxybutane, enabling thus the expansion of studies on the biological properties of these compounds.
Experimental
General. Melting points were measured on a Boëtius hot plate microscope and are uncorrected. The IR spectra were recorded on a Nicolet Impact 410 spectrometer, in KBr pellets. The high performance liquid chromatography (HPLC) analyses were performed with an Agilent Chromatograph 1200 Series at room temperature by isocratic elution of acrylonitrile on an Agilent Zorbax SB-C18 (250 × 4.6) column with a flow rate of 1.0 mL/min. The NMR spectra have been recorded on a Bruker Avance III 400 instrument operating at 400.1, 100.6 and 40.6 MHz for 1H, 13C, and 15N nuclei respectively. Samples were transferred in 5 mm Wilmad 507 NMR tubes and recorded with either a 5 mm multinuclear inverse detection z-gradient probe (1H spectra and all H,H/H,C/H,N 2D experiments) or with a 5 mm four nuclei direct detection z-gradient probe for 13C spectra. Chemical shifts are reported in δ units (ppm) and were referenced to internal TMS for 1H nuclei, to the internal deuterated solvent for 13C nuclei (CDCl3 referenced at 77.0 ppm), and referenced to liquid ammonia (0.0 ppm) using nitromethane (380.2 ppm) as external standard for 15N nuclei. Unambiguous 1D NMR signal assignments were made based on 2D NMR homo- and heteronuclearcorrelations. H,H-COSY, H,H-NOESY, H,C-HSQC and H,C-HMBC experiments were recorded using standard pulse sequences in the version with z-gradients, as delivered by Bruker with TopSpin 2.1 PL6 spectrometer control and processing software. H,C-undecoupled-HSQC experiments have been recorded using the pulse sequence described by S. Simova [21]. The 15N chemical shifts were obtained as projections from the 2D indirectly detected H,N-HMBC spectra, employing a standard pulse sequence in the version with z-gradients as delivered by Bruker (TopSpin 2.1 PL6). Elemental analyses for C, H and N were obtained using a COSTECH Instruments EAS32. Satisfactory microanalyses for all new compounds were obtained.
Benzimidazole, 5,6-dimethylbenzimidazole, activated acetylenic esters, 3-butyn-2-one and alkyl bromoacetates were purchased from Aldrich and used without further purification. 1-Benzylbenzimidazole, 1-benzyl-5,6-dimethylbenzimidazole and 1-ethyl-5,6-dimethylbenzimidazole were obtained from corresponding benzimidazoles and benzyl chloride, respectively ethyl bromide. 1-Benzylbenzimidazolium bromides (6) were obtained from 1-benzylbenzimidazole, respectively 1-benzyl-5,6-dimethylbenzimidazole, and alkyl bromoacetate in acetone, according previously reported methods [8]. Tetrapyridinecobalt(II) dichromate (TPCD) was obtained according the reported method [22].
General procedure for the reaction of 1-substituted benzimidazoles (1a–c) with ethyl bromoacetate (2) and non-symmetrical alkynes (3a–c) in 1,2-epoxybutane. A mixture of 1-substituted benzimidazole 1a–c (2 mmol), ethyl bromoacetate 2 (2 mmol) and an alkyne 3a–c (2 mmol) in 30 mL of 1,2-epoxybutane was heated at reflux temperature (approx. 62 °C) for 24 hours. The solvent was partly removed under vacuum, 3 mL of MeOH was added under a gentle stirring, and the mixture was left 2 hours in the refrigerator. The solid formed was filtered off and recrystallized from MeOH/Et2O giving pyrrolo[1,2-a]quinoxalin-4-one 4a–g. The filtrate was concentrated under vacuum and chromatographed on a SiO2 packed column by eluting with EtOAc:hexane (1:4 v/v) giving pyrrolo[1,2-a]benzimidazole 5 and an additional quantity of pyrrolo[1,2-a]quinoxalin-4-one 4 (the order of elution: 4<5).
Ethyl 4-oxo-5-benzylpyrrolo[1,2-a]quinoxalin-2-carboxylate (4b). 0.29 g (42%) pale yellow crystals. FTIR (νmax, cm−1): 3121, 2975, 1710, 1651, 1611, 1551, 1519, 1426, 1361, 1305, 1270, 1196, 1165, 1096, 1023; 1H NMR (CDCl3) δ 1.41 (3H, t, 7.2 Hz, CH3), 4.38 (2H, quartet, 7.2 Hz, CH2), 5.50 (2H, bs, CH2), 7.19–7.33 (8H, m, aromatic rings), 7.68 (1H, d, 1.6 Hz, H-3), 7.72–7.73 (1H, m, H-9), 8.24 (1H, d, 1.6 Hz, H-1). The individual chemical shifts, multiplicities and coupling constants for the 7.19–7.33 multiplet were obtained from undecoupled HSQC as follows: 7.21 (1H, m, H-8), 7.236 (1H, t, 8.1 Hz, H-7), 7.239 (1H, d, 8.2 Hz, H-6), 7.25 (1H, t, 7.4 Hz, H-4’), 7.28 (2H, d, 7.2 Hz, H-2’), 7.31 (2H, t, 7.3 Hz, H-3’) ppm; 13C NMR (CDCl3) δ 14.38 (CH3), 45.12 (CH2), 60.57 (OCH2), 113.97 (C-3), 114.99 (C-9), 116.88 (C-6), 119.42 (C-1), 120.43 (C-2), 123.27 (C-8), 123.37 (C-9a), 123.59 (C-3a), 126.58 (C-2’), 126.78 (C-7), 127.45 (C-4’), 128.87 (C-3’), 129.97 (C-5a), 136.04 (C-1’), 155.48 (C-4), 163.77 (COO); 15N NMR (CDCl3) δ 136.4 (N-5), 173.5 (N-10) ppm; anal. calcd for C21H18N2O3 (346.38): C, 72.82; H, 5.24; N, 8.09%; found: C, 72.90; H, 5.31; N, 8.01%.
Diethyl 4-benzyl-4H-pyrrolo[1,2-a]benzimidazole-1,3-dicarboxylate (5b). 0.1 g (13%) pale yellow crystals. FTIR (νmax, cm−1): 1700, 1685, 1580, 1514, 1479, 1453, 1400, 1303, 1290, 1233, 1181, 1136, 1106, 1070; 1H NMR (CDCl3) δ 1.37 (3H, t, 7.2 Hz, CH3-3), 1.48 (3H, t, 7.2 Hz, CH3-1), 4.32 (2H, quartet, 7.2 Hz, CH2-3), 4.45 (2H, quartet, 7.2 Hz, CH2-1), 6.13 (2H, bs, CH2), 7.25–7.32 (8H, m, aromatic rings), 7.78 (1H, s, H-2), 8.88 (1H, d, 8.2 Hz, H-8). The individual chemical shifts, multiplicities and coupling constants for the 7.25–7.32 multiplet were obtained from undecoupled HSQC as follows: 7.240 (2H, d, 7.5 Hz, H-2’), 7.248 (1H, t, 7.3 Hz, H-4’), 7.25 (1H, d, 8.2 Hz, H-5), 7.26 (1H, t, 8 Hz, H-7), 7.29 (2H, t, 7.4 Hz, H-3’), 7.30 (1H, t, 8 Hz, H-6) ppm; 13C NMR (CDCl3) δ 14.47 (CH3-3), 14.59 (CH3-1), 48.48 (CH2), 59.93 (CH2-3), 60.24 (CH2-1), 91.75 (C-3), 110.20 (C-5), 112.32 (C-1), 116.23 (C-8), 121.37 (C-7), 124.14 (C-6), 125.20 (C-2), 126.79 (C-2’), 127.07 (C-8a), 127.57 (C-4’), 128.73 (C-3’), 136.25 (C-4a), 136.91 (C-1’), 143.08 (C-3a), 160.68 (COO-1), 163.63 (COO-3) ppm; 15N NMR (CDCl3) δ 116.9 (N-4), 172.1 (N-9) ppm; anal. calcd. for C23H22N2O4 (390.43): C, 70.75; H, 5.68; N, 7.17%; found: C, 70.67; H, 5.61; N, 7.23%.
General procedure for the reaction of 1-benzylbenzimidazolium bromides (6) with DMAD (3d) in 1,2-epoxybutane. A mixture of a 1-benzylbenzimidazolium bromide 6 (2 mmol) and DMAD 3d (2 mmol) in 30 mL of 1,2-epoxybutane was heated at reflux temperature for 24 hours. The solvent was removed under vacuum, and the residue was chromatographed on a SiO2 packed column by eluting with EtOAc:hexane (1:4 v/v) giving pyrrolo[1,2-a]quinoxalin-4-ones 4h,i and the pyrrolo[1,2-a]benzimidazole 5h (the order of elution: 4<5).
Dimethyl 4-oxo-5-benzylpyrrolo[1,2-a]quinoxalin-2,3-dicarboxylate (4h). 0.33 g (42%) white crystals. FTIR (νmax, cm–1): 1748, 1710, 1663, 1523, 1412, 1370, 1270, 1246, 1198, 1153, 1074; 1H NMR (CDCl3) δ 3.90 (3H, s, CH3-2), 4.05 (3H, s, CH3-3), 5.47 (2H, bs, CH2), 7.22–7.33 (8H, m, aromatic rings), 7.72–7.74 (1H, m, H-9), 8.19 (1H, s, H-1). The individual chemical shifts, multiplicities and coupling constants for the 7.22–7.33 multiplet were obtained from undecoupled HSQC as follows: 7.24 (1H, t, 7.7 Hz, H-8), 7.25 (1H, t, 7.2 Hz, H-4’), 7.254 (1H, d, 8.7 Hz, H-6), 7.26 (2H, d, 7.9 Hz, H-2’), 7.28 (1H, t, 7.9 Hz, H-7), 7.31 (2H, t, 7.72 Hz, H-3’); 13C NMR (CDCl3) δ 45.20 (CH2), 52.10 (CH3-2), 53.12 (CH3-3), 115.18 (C-9), 117.07 (C-6), 117.85 (C-2), 118.81 (C-1), 120.80 (C-3a), 121.21 (C-3), 122.66 (C-9a), 123.58 (C-8), 126.58 (C-2’), 127.46 (C-7), 127.55 (C-4’), 128.91 (C-3’), 129.92 (C-5a), 135.57 (C-1’), 154.43 (C-4), 162.84 (COO-2), 165.42 (COO-3) ppm; 15N NMR (CDCl3) δ 137.6 (N-5), 172.0 (N-10) ppm; anal. calcd for C22H18N2O5 (390.39): C, 67.68; H, 4.65; N, 7.18%; found: C, 67.75; H, 4.68; N, 7.12%.
Dimethyl 1-carbethoxy-4-benzyl-4H-pyrrolo[1,2-a]benzimidazole-2,3-dicarboxylate (5h). 0.14 g (16%) pale yellow crystals. FTIR (νmax, cm−1): 2997, 2951, 1745, 1710, 1687, 1663, 1572, 1522, 1456, 1408, 1369, 1269, 1216, 1177, 1140, 1066, 1074. 1H NMR (CDCl3) δ 1.44 (3H, t, 7.2 Hz, CH3-Et), 3.81 (3H, s, CH3-3), 4.01 (3H, s, CH3-2), 4.43 (2H, quartet, 7.2 Hz, CH2-Et), 6.08 (2H, bs, CH2), 7.22–7.38 (8H, m, aromatic rings), 8.86 (1H, d, 8.0 Hz, H-8). The individual chemical shifts, multiplicities and coupling constants for the 7.22–7.38 multiplet were obtained from undecoupled HSQC as follows: 7.22 (2H, d, 7.6 Hz, H-2’), 7.27 (1H, t, 7.5 Hz, H-4’), 7.29 (1H, d, 8.3 Hz, H-5), 7.30 (1H, t, 8.1 Hz, H-7), 7.31 (2H, t, 7.6 Hz, H-3’), 7.35 (1H, t, 8 Hz, H-6) ppm; 13C NMR (CDCl3) δ 14.21 (CH3-Et), 48.51 (CH2), 51.58 (CH3-3), 52.58 (CH3-2), 60.88 (CH2-Et), 89.98 (C-3), 109.71 (C-1), 110.29 (C-5), 116.62 (C-8), 121.75 (C-7), 124.79 (C-6), 126.58 (C-2’), 126.63 (C-8a), 127.66 (C-4’), 128.78 (C-3’), 130.49 (C-2), 136.41 (C-4a), 136.57 (C-1), 141.86 (C-3a), 159.58 (COO-Et), 162.76 (COO-3), 166.10 (COO-2) ppm; 15N NMR (CDCl3) δ 116.1 (N-4), 168.7 (N-9) ppm; anal. calcd for C24H22N2O6 (434.44): C, 66.35; H, 5.10; N, 6.45%; found: C, 66.31; H, 5.14; N, 6.39%.
General synthetic procedure for pyrrolo[1,2-a]quinoxalin-4-ones 10a–f. A mixture of a benzimidazole 9 (2 mmol), alkyl bromoacetate 2 (4 mmol) and a non-symmetrical alkyne 3 (2 mmol) in 30 mL of 1,2-epoxybutane was heated at reflux temperature for 30 hours. The solvent was partly removed under vacuum, 3 mL of MeOH was added under a gentle stirring, and the mixture was left over night in a refrigerator. The formed solid was filtered off and recrystallized from MeOH giving pyrrolo[1,2-a]quinoxalin-4-one 10a–f.
Ethyl 2-(2-acetyl-4-oxo-pyrrolo[1,2-a]quinoxalin-5-yl) acetate (10a). 0.235 g (38%) beige crystals, mp 193–194 °C. FTIR (νmax, cm−1): 3109, 2984, 1746, 1656, 1617, 1549, 1516, 1420, 1383, 1357, 1277, 1206; 1H NMR (CDCl3) δ 1.27 (3H, t, 7.2 Hz, CH3-Et), 2.56 (3H, s, CH3), 4.25 (2H, quartet, 7.2 Hz, CH2-Et), 5.04 (2H, s, CH2), 7.08 (1H, d, 8.3 Hz, H-6), 7.28 (1H, t, 7.2 Hz, H-8), 7.36 (1H, t, 7.3 Hz, H-7), 7.59 (1H, d, 1.5 Hz, H-3), 7.75 (1H, d, 8.1 Hz, H-9), 8.21 (1H, d, 1.5 Hz, H-1) ppm; 13C NMR (CDCl3) δ 14.13 (CH3-Et), 27.66 (CH3), 42.83 (CH2), 61.93 (CH2-Et), 113.52 (C-3), 115.39 (C-9), 115.45 (C-6), 118.55 (C-1), 123.19 (C-9a), 123.50 (C-3a), 123.69 (C-8), 127.18 (C-7), 128.48 (C-2), 129.94 (C-5a), 155.07 (C-4), 167.89 (COO), 193.65 (CO)ppm; 15N NMR (CDCl3) δ 129.9 (N-5), 175.3 (N-10) ppm; anal. calcd for C17H16N2O4 (312.32): C, 65.37; H, 5.16; N, 8.97%; found: C, 65.48; H, 5.20; N, 8.88%.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, 1H, 13C and 15N NMR spectra for all new compounds. | ||
| Format: PDF | Size: 202.8 KB | Download |
Acknowledgements
This work was supported by a grant of the Romanian National Authority for Scientific Research, CNCS-UEFISCDI, project number PN-II-ID-PCCE-2011-2-0028. The Paper is dedicated to the 65th anniversary of “Petru Poni” Institute of Macromolecular Chemistry of the Romanian Academy, Iasi, Romania.
References
-
Guillon, J.; Reynolds, R. C.; Leger, J.-M.; Guie, M. A.; Massip, S.; Dallemagne, P.; Jarry, C. J. Enzyme Inhib. Med. Chem. 2004, 19, 489–495. doi:10.1080/14756360412331280464
Return to citation in text: [1] -
Maga, G.; Gemma, S.; Fattorusso, C.; Locatelli, G. A.; Butini, S.; Persico, M.; Kukreja, G.; Romano, M. P.; Chiasserini, L.; Savini, L.; Novellino, E.; Nacci, V.; Spadari, S.; Campiani, G. Biochemistry 2005, 44, 9637–9643. doi:10.1021/bi047437u
Return to citation in text: [1] -
Desplat, V.; Geneste, A.; Begorre, M.-A.; Fabre, S. B.; Brajot, S.; Massip, S.; Thiolat, D.; Mossalayi, D.; Jarry, C.; Guillon, J. J. Enzyme Inhib. Med. Chem. 2008, 23, 648–653. doi:10.1080/14756360802205448
Return to citation in text: [1] -
Sabatucci, J. P.; Mahaney, P. E. Pyrrolo[1,2-a]quinoxalin-5-(4H)-yl)sulfonyls and Carbonyls and their Use as Estrogenic Agents. PCT Int. Appl. WO 2006/068928 A1, June 29, 2006.
Return to citation in text: [1] -
Kalinin, A. A.; Mamedov, V. A. Chem. Heterocycl. Compd. 2011, 46, 1423–1442. doi:10.1007/s10593-011-0688-1
Return to citation in text: [1] -
Mamedov, V. A.; Kalinin, A. A. Chem. Heterocycl. Compd. 2010, 46, 641–664. doi:10.1007/s10593-010-0565-3
Return to citation in text: [1] -
Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010
Return to citation in text: [1] [2] [3] [4] [5] -
Zugravescu, I.; Herdan, J.; Druta, I. Rev. Roum. Chim. 1974, 19, 649–658.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Meth-Cohn, O. Tetrahedron Lett. 1975, 16, 413–416. doi:10.1016/S0040-4039(00)71880-6
Return to citation in text: [1] [2] [3] -
Wu, K.; Chen, Q.-Y. Synthesis 2003, 35–40. doi:10.1055/s-2003-36246
Return to citation in text: [1] [2] [3] -
Fang, X.; Wu, Y.-M.; Deng, J.; Wong, S.-W. Tetrahedron 2004, 60, 5487–5493. doi:10.1016/j.tet.2004.04.012
Return to citation in text: [1] [2] [3] -
Wang, B.; Hu, J.; Zhang, X.; Hu, Y.; Hu, H. J. Heterocycl. Chem. 2000, 37, 1533–1537. doi:10.1002/jhet.5570370620
Return to citation in text: [1] [2] [3] [4] -
Matsuda, Y.; Yamashita, K.; Takahashi, K.; Ide, S.; Torisu, K.; Furuno, K. Heterocycles 1992, 33, 295–302. doi:10.3987/COM-91-S35
Return to citation in text: [1] [2] -
Piltan, M.; Moradi, L.; Abasi, G.; Zarei, S. A. Beilstein J. Org. Chem. 2013, 9, 510–515. doi:10.3762/bjoc.9.55
Return to citation in text: [1] -
Huang, A.; Liu, F.; Zhan, C.; Liu, Y.; Ma, C. Org. Biomol. Chem. 2011, 9, 7351–7357. doi:10.1039/c1ob05936j
Return to citation in text: [1] -
Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036
Return to citation in text: [1] [2] -
Georgescu, E.; Caira, M. R.; Georgescu, F.; Drăghici, B.; Popa, M. M.; Dumitrascu, F. Synlett 2009, 1795–1799. doi:10.1055/s-0029-1217367
Return to citation in text: [1] -
Dumitrascu, F.; Caproiu, M. T.; Georgescu, F.; Draghici, B.; Popa, M. M.; Georgescu, E. Synlett 2010, 2407–2410. doi:10.1055/s-0030-1258035
Return to citation in text: [1] -
Caira, M. R.; Georgescu, E.; Georgescu, F.; Albota, F.; Dumitrascu, F. Monatsh. Chem. 2011, 142, 743–748. doi:10.1007/s00706-011-0468-8
Return to citation in text: [1] -
Georgescu, E.; Georgescu, F.; Popa, M. M.; Draghici, C.; Tarko, L.; Dumitrascu, F. ACS Comb. Sci. 2012, 14, 101–107. doi:10.1021/co2002125
Return to citation in text: [1] -
Simova, S. Magn. Reson. Chem. 1998, 36, 505–510. doi:10.1002/(SICI)1097-458X(199807)36:7<505::AID-OMR300>3.0.CO;2-H
Return to citation in text: [1] -
Wei, X.; Hu, Y.; Li, T.; Hu, H. J. Chem. Soc., Perkin Trans. 1 1993, 2487–2489. doi:10.1039/p19930002487
Return to citation in text: [1]
| 21. | Simova, S. Magn. Reson. Chem. 1998, 36, 505–510. doi:10.1002/(SICI)1097-458X(199807)36:7<505::AID-OMR300>3.0.CO;2-H |
| 22. | Wei, X.; Hu, Y.; Li, T.; Hu, H. J. Chem. Soc., Perkin Trans. 1 1993, 2487–2489. doi:10.1039/p19930002487 |
| 1. | Guillon, J.; Reynolds, R. C.; Leger, J.-M.; Guie, M. A.; Massip, S.; Dallemagne, P.; Jarry, C. J. Enzyme Inhib. Med. Chem. 2004, 19, 489–495. doi:10.1080/14756360412331280464 |
| 5. | Kalinin, A. A.; Mamedov, V. A. Chem. Heterocycl. Compd. 2011, 46, 1423–1442. doi:10.1007/s10593-011-0688-1 |
| 11. | Fang, X.; Wu, Y.-M.; Deng, J.; Wong, S.-W. Tetrahedron 2004, 60, 5487–5493. doi:10.1016/j.tet.2004.04.012 |
| 4. | Sabatucci, J. P.; Mahaney, P. E. Pyrrolo[1,2-a]quinoxalin-5-(4H)-yl)sulfonyls and Carbonyls and their Use as Estrogenic Agents. PCT Int. Appl. WO 2006/068928 A1, June 29, 2006. |
| 12. | Wang, B.; Hu, J.; Zhang, X.; Hu, Y.; Hu, H. J. Heterocycl. Chem. 2000, 37, 1533–1537. doi:10.1002/jhet.5570370620 |
| 3. | Desplat, V.; Geneste, A.; Begorre, M.-A.; Fabre, S. B.; Brajot, S.; Massip, S.; Thiolat, D.; Mossalayi, D.; Jarry, C.; Guillon, J. J. Enzyme Inhib. Med. Chem. 2008, 23, 648–653. doi:10.1080/14756360802205448 |
| 9. | Meth-Cohn, O. Tetrahedron Lett. 1975, 16, 413–416. doi:10.1016/S0040-4039(00)71880-6 |
| 2. | Maga, G.; Gemma, S.; Fattorusso, C.; Locatelli, G. A.; Butini, S.; Persico, M.; Kukreja, G.; Romano, M. P.; Chiasserini, L.; Savini, L.; Novellino, E.; Nacci, V.; Spadari, S.; Campiani, G. Biochemistry 2005, 44, 9637–9643. doi:10.1021/bi047437u |
| 16. | Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036 |
| 14. | Piltan, M.; Moradi, L.; Abasi, G.; Zarei, S. A. Beilstein J. Org. Chem. 2013, 9, 510–515. doi:10.3762/bjoc.9.55 |
| 15. | Huang, A.; Liu, F.; Zhan, C.; Liu, Y.; Ma, C. Org. Biomol. Chem. 2011, 9, 7351–7357. doi:10.1039/c1ob05936j |
| 16. | Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036 |
| 7. | Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010 |
| 8. | Zugravescu, I.; Herdan, J.; Druta, I. Rev. Roum. Chim. 1974, 19, 649–658. |
| 7. | Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010 |
| 8. | Zugravescu, I.; Herdan, J.; Druta, I. Rev. Roum. Chim. 1974, 19, 649–658. |
| 9. | Meth-Cohn, O. Tetrahedron Lett. 1975, 16, 413–416. doi:10.1016/S0040-4039(00)71880-6 |
| 10. | Wu, K.; Chen, Q.-Y. Synthesis 2003, 35–40. doi:10.1055/s-2003-36246 |
| 11. | Fang, X.; Wu, Y.-M.; Deng, J.; Wong, S.-W. Tetrahedron 2004, 60, 5487–5493. doi:10.1016/j.tet.2004.04.012 |
| 12. | Wang, B.; Hu, J.; Zhang, X.; Hu, Y.; Hu, H. J. Heterocycl. Chem. 2000, 37, 1533–1537. doi:10.1002/jhet.5570370620 |
| 13. | Matsuda, Y.; Yamashita, K.; Takahashi, K.; Ide, S.; Torisu, K.; Furuno, K. Heterocycles 1992, 33, 295–302. doi:10.3987/COM-91-S35 |
| 6. | Mamedov, V. A.; Kalinin, A. A. Chem. Heterocycl. Compd. 2010, 46, 641–664. doi:10.1007/s10593-010-0565-3 |
| 7. | Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010 |
| 13. | Matsuda, Y.; Yamashita, K.; Takahashi, K.; Ide, S.; Torisu, K.; Furuno, K. Heterocycles 1992, 33, 295–302. doi:10.3987/COM-91-S35 |
| 17. | Georgescu, E.; Caira, M. R.; Georgescu, F.; Drăghici, B.; Popa, M. M.; Dumitrascu, F. Synlett 2009, 1795–1799. doi:10.1055/s-0029-1217367 |
| 18. | Dumitrascu, F.; Caproiu, M. T.; Georgescu, F.; Draghici, B.; Popa, M. M.; Georgescu, E. Synlett 2010, 2407–2410. doi:10.1055/s-0030-1258035 |
| 19. | Caira, M. R.; Georgescu, E.; Georgescu, F.; Albota, F.; Dumitrascu, F. Monatsh. Chem. 2011, 142, 743–748. doi:10.1007/s00706-011-0468-8 |
| 20. | Georgescu, E.; Georgescu, F.; Popa, M. M.; Draghici, C.; Tarko, L.; Dumitrascu, F. ACS Comb. Sci. 2012, 14, 101–107. doi:10.1021/co2002125 |
| 12. | Wang, B.; Hu, J.; Zhang, X.; Hu, Y.; Hu, H. J. Heterocycl. Chem. 2000, 37, 1533–1537. doi:10.1002/jhet.5570370620 |
| 7. | Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010 |
| 8. | Zugravescu, I.; Herdan, J.; Druta, I. Rev. Roum. Chim. 1974, 19, 649–658. |
| 7. | Ogura, H.; Kikuchi, K. J. Org. Chem. 1972, 37, 2679–2682. doi:10.1021/jo00982a010 |
| 10. | Wu, K.; Chen, Q.-Y. Synthesis 2003, 35–40. doi:10.1055/s-2003-36246 |
| 11. | Fang, X.; Wu, Y.-M.; Deng, J.; Wong, S.-W. Tetrahedron 2004, 60, 5487–5493. doi:10.1016/j.tet.2004.04.012 |
| 12. | Wang, B.; Hu, J.; Zhang, X.; Hu, Y.; Hu, H. J. Heterocycl. Chem. 2000, 37, 1533–1537. doi:10.1002/jhet.5570370620 |
| 9. | Meth-Cohn, O. Tetrahedron Lett. 1975, 16, 413–416. doi:10.1016/S0040-4039(00)71880-6 |
© 2014 Georgescu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)