Abstract
The marine natural product malevamide D from the cyanobacterium Symploca hydnoides was synthesized for the first time. The final peptide coupling linked the dolaisoleuine and dolaproine subunits. The phenyl group of malevamide D was also functionalized with a photoreactive diazirine moiety, which was carried through seven reaction steps. Comprehensive assessment of the cytotoxicity in a panel of 42 human cancer cell lines revealed a geomean IC70 value of 1.5 nM (IC50 0.7 nM) for malevamide D, whereas the photoreactive derivative proved to be less active by a factor of at least 200. COMPARE analysis indicated tubulin interaction as likely mode of action of malevamide D.

Graphical Abstract
Introduction
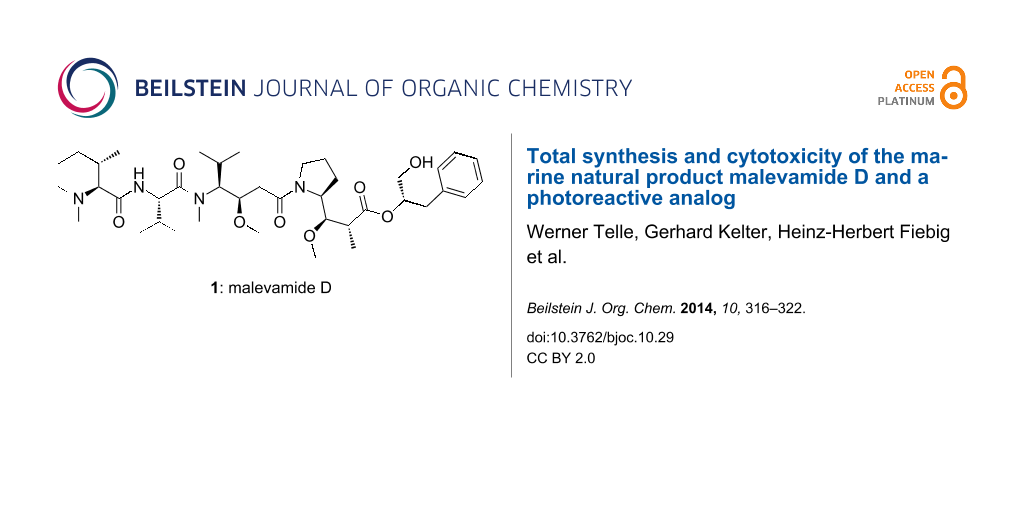
The depsipeptide malevamide D (1, Figure 1) belongs to the dolastatin class of marine natural products and has been isolated from the cyanobacterium Symploca hydnoides by Scheuer and co-workers in 2002 (7.5 mg, 0.014% of the dry weight) [1]. Malevamide D (1) was reported to exhibit in vitro cytotoxicity in the subnanomolar range (IC50 values about 0.7 nM). Closely related to malevamide D (1) is isodolastatin H (2), which is also active in vivo, but slightly weaker than dolastatin 10 (3) [2]. Although there has been no study regarding the mode of action, it can be assumed that malevamide D (1) acts similarly to dolastatin 10 (3) by inhibiting the formation of microtubuli from tubulin and, thereby, cell division. Dolastatin-type natural products continue to be tested in clinical trials for cancer therapy [3]. This includes an antibody conjugate of monomethylauristatin E (MMAE), which has been approved for the treatment of Hodgkin lymphoma (Brentuximab vedotin) [4].
![[1860-5397-10-29-1]](/bjoc/content/figures/1860-5397-10-29-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the strongly cytotoxic marine natural products malevamide D (1), isodolastatin H (2), and dolastatin 10 (3).
Figure 1: Structures of the strongly cytotoxic marine natural products malevamide D (1), isodolastatin H (2),...
In this paper, we report the first total synthesis of malevamide D (1) and an assessment of its in vitro cytotoxicity in a panel of 42 cell lines. We also synthesized a diazirinylated analog of 1, which, by photocrosslinking, might contribute to the understanding of the binding of 1 to tubulin.
Results and Discussion
Total synthesis of malevamide D
The natural products malevamide D (1), isodolastatin H (2), and dolastatin 10 (3) share two β-methoxy-γ-amino acid building blocks. The closest structural analog of malevamide D (1) is isodolastatin H (2) with an identical ester section composed of dolaproine and (2S)-3-phenylpropan-1,2-diol. On the N-terminal side, the isopropyl and two sec-butyl side chains of isodolastatin H (2) are replaced by one sec-butyl and two isopropyl side chains, respectively, in the case of malevamide D (1).
The total synthesis of 1 adopts the strategy by Yamada and co-workers in their total synthesis of 2, which sets the first retro cut at the tertiary amide bond between dolaisoleuine and dolaproine [2]. The tert-butyl ester 7 of Cbz-protected 3-methoxy-5-methyl-4-(methylamino)hexanoic acid (MMMAH) was obtained in multigram quantities within five steps from Cbz-valine (8), adapting a convenient sequence developed for dolaisoleuine by Pettit and co-workers [5]. Cbz-protected N-methylvalinol (4) [6] was oxidized to the aldehyde 5 under Parikh–Doering conditions, followed by aldol addition of the lithium enolate of t-BuOAc (Scheme 1). β-Hydroxyhexanoate 6 was obtained as a mixture of diastereomers (5:4), which was separated by column chromatography (silica) on a multigram scale. Treatment of (3R,4S)-6 with Meerwein's salt in presence of proton sponge afforded Cbz-protected MMMAH tert-butyl ester 7 in 87% yield. There are also other, enantioselective syntheses of 7 [7] and Boc-protected versions of MMMAH [8,9], but we intended to also have the (3S)-epimer of 7 at our disposal.
![[1860-5397-10-29-i1]](/bjoc/content/inline/1860-5397-10-29-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Total synthesis of malevamide D (1). a) DMSO (16 equiv), NEt3 (5 equiv), pyridine·SO3 (5 equiv), 0 °C, 1 h, 91%. b) LDA (2.5 equiv), t-BuOAc (1.3 equiv), THF, −78 °C, 1 h; separation of diastereomers, (3R,4S)-6: 47%. c) 1,8-bis(dimethylamino)naphthalene (2.5 equiv), Me3OBF4 (2.6 equiv), 4 Å MS, 1,2-DCE, 0 °C, 2 h, rt, 16 h, 87%. d) H2, Pd/C (5%), MeOH, rt, 1 h. e) 8 (3 equiv), NEt3 (4 equiv), DEPCl (2 equiv), DCM, 0 °C, 2 h, rt, 16 h, 80% from 7. f) H2, Pd/C (5%), MeOH, rt, 90 min. g) 10 (3 equiv), NEt3 (4 equiv), DEPC (1.5 equiv), DCM, 0 °C, 2 h, rt, 16 h, 83% from 9. h) TFA, DCM, 0 °C, 90 min, quant. i) 13 (1 equiv), NEt3 (6 equiv), DEPC (1.3 equiv), DCM, 0 °C to rt, 16 h, 68%. j) TBAF·3H2O (5 equiv), THF/H2O 19:1, rt, 3 h, 71%. DEPCl: diethyl phosphorochloridate; DEPC: diethyl phosphorocyanidate.
Scheme 1: Total synthesis of malevamide D (1). a) DMSO (16 equiv), NEt3 (5 equiv), pyridine·SO3 (5 equiv), 0 ...
After hydrogenation of 7, the resulting secondary amine was coupled with Cbz-valine (8) affording dipeptide 9 (DEPCl, diethyl phosphorochloridate), 80% over two steps, Scheme 1). N,N-Dimethylisoleucine (10), obtained by reductive dimethylation of isoleucine with aqueous formaldehyde and H2 on Pd/C [10,11], was appended at the N-terminus (DEPC, diethyl phosphorocyanidate) affording tripeptide tert-butyl ester 11. Treatment with TFA liberated the free acid 12, which was coupled with the trifluoroacetate of dolaproine ester 13, liberated from the N-Boc-protected analog [2].
With the subsequent coupling step affording TBDPS-protected malevamide D (14), we initially had difficulties. It proved to be crucial to remove excess of TFA from both coupling partners 12 and 13 completely, by repeated evaporation of DCM solutions. Finally, a coupling yield of 68% was reached following the DEPC protocol. Desilylation (TBAF in THF/H2O) afforded 1 (HRESIMS found 755.49257, calcd. for C40H68NaN4O8 755.49294) in 12% yield after 10 steps from valinol 4 (longest linear sequence). The optical rotation of the synthesized material ([α]D28 −37, c 0.09, MeOH) is a little smaller than reported for the isolated natural product ([α]D26 −55, c 0.10, MeOH) [1]. The 1H and 13C NMR spectra of 1 shows two sets of signals in a ratio of ca. 1:1. Clearly separated in the 13C NMR spectrum are, for instance, the ester carbonyl signals of the conformers (174.0 and 173.3 ppm), the carbonyl signals of the pyrrolidine amide (170.5 and 170.6 ppm), and all phenyl carbon signals. Of the eastern section, only three of the four pyrrolidine carbon signals are broad. Most of the carbon signals of the western tripeptide of malevamide D are broadened (see Supporting Information File 1). The 1H NMR spectrum shows many broad and overlapping signals, the assignment of which was only possible by careful interpretation of the 2D NMR data set. The only isolated signals belonging to different signal sets are located at δH 3.84 (one of the diastereotopic OCH2 signals) and 5.21 ppm (acylated carbinol). The two sets of signals are probably caused by the presence of both conformers of the acyl pyrrolidine. An assignment of the data sets to either of the two conformers was not possible. The occurrence of conformers has been mentioned by Scheuer and co-workers and is also known for dolastatin 10 [12] and symplostatin 1 [13].
As an alternative route to the N-terminal tripeptide 11, we also investigated the coupling of the N-terminal dipeptide 15 with MMMAH tert-butyl ester (16, Scheme 2). However, after treatment of dipeptide 15 with secondary amine 16 (0.43 equiv) and DEPC (17, 2.5 equiv), the only isolable product was oxazolylphosphate 18 (35%) without any amide formation occurring. The structure elucidation of 18 required C–P coupling constant analysis, which showed doublet 13C NMR signals of oxazole C-5 (10 Hz) and C-4 (6.2 Hz). To the best of our knowledge, this is only the second time that a peptide-derived oxazol-5-ylphosphate has been characterized. Earlier, Boyd and co-workers had converted an oxazolone to an oxazolyl phosphate by treatment with excess diphenyl chlorophosphate [14]. In our case, a similar pathway is to be assumed with the oxazolone being formed first. The secondary amine 16 appears to react too slowly to be able to compete with oxazolone formation.
![[1860-5397-10-29-i2]](/bjoc/content/inline/1860-5397-10-29-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of oxazolylphosphate 18 on attempted DEPC-mediated coupling of dipeptide 15.
Scheme 2: Formation of oxazolylphosphate 18 on attempted DEPC-mediated coupling of dipeptide 15.
Diazirinyl-substituted malevamide D
Encouraged by the strong cytotoxicity of 1, we addressed the synthesis of a photoactivatable analog. The question was whether such a derivative would still be cytotoxic. The phenyl ring of 1 was chosen as location of a trifluoromethyldiazirinyl substituent. Pettit and co-workers have shown that introduction of a phosphate moiety at the p-position of the phenyl ring of auristatin PE did not lead to loss of cytotoxicity [3]. Recently, we have synthesized a hemiasterlin derivative with a diazirinylated indole moiety by incorporation of a thermally stable L-phototryptophan carrying a 1-azi-2,2,2-trifluoroethyl unit [15].
Racemic photo phenylpropanediol 25 was obtained starting from aryl(trifluoromethyl)ketone 19 (Scheme 3), which itself was synthesized via Grignard monoallylation of 1,4-dibromobenzene, followed by dihydroxylation, dioxolane protection and trifluoroacetylation of the remaining brominated position [16]. Refluxing of ketone 19 with NH2OH·HCl in pyridine led to nearly quantitative hydrolysis of the dioxolane affording diol oxime 20 as 95:5 mixture of E- and Z-isomers. However, if solid NaOH was added to the reaction mixture and heated for several hours, the dioxolane survived, affording a 1:1 mixture of E/Z-oximes, which were tosylated after short work-up to (E)- and (Z)-22.
![[1860-5397-10-29-i3]](/bjoc/content/inline/1860-5397-10-29-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of tosyloximes (Z)-22 and (E)-22, X-ray structure of (E)-22. a) NH2OH·HCl (1.5 equiv), pyridine, reflux, 2 h, 80%. b) 2,2-dimethoxypropane (12.4 equiv), p-TsOH·H2O (5 mol %), DMF, rt, 1 h, 95%. c) p-TsCl (1.05 equiv), NEt3 (1.3 equiv), DCM, 0 °C to rt, 18 h, 83%. d) (i) NH2OH·HCl, NaOH, EtOH, pyridine, reflux (for details see Supporting Information File 1); (ii) p-TsCl (1.05 equiv), NEt3 (1.3 equiv), DCM, 0 °C to rt, 18 h, 74%.19F NMR chemical shifts (ppm) in CDCl3.
Scheme 3: Synthesis of tosyloximes (Z)-22 and (E)-22, X-ray structure of (E)-22. a) NH2OH·HCl (1.5 equiv), py...
Reprotection of the diol unit of 20 and tosylation of 21 afforded tosyloxime (E)-22, which crystallized overnight from the neat mixture and was analyzed by X-ray crystallography [17]. Thus, it became possible to assign the 19F NMR chemical shifts of both isomers ((E)-22: δF −66.9; (Z)-22: δF −61.9).
Conversion of (E)-22 to diaziridine 23 (colorless solid, δF −75.96, −75.93) was performed quantitatively (liquid NH3 in t-BuOMe, Scheme 4). As expected, the NMR spectra of the colorless solid 23 showed two sets of signals. Oxidation of 23 with I2/NEt3 to diazirine 24 (δF −65.7) and acidic deprotection afforded diazirine diol 25. Figure 2 shows the DSC (differential scanning calorimetry) profile of 25, which melts at 54 °C and starts to decompose above 80 °C, indicating sufficient thermal stability for biological studies. Monosilylation of the primary hydroxy group of 25 afforded building block 26. Coupling with Boc-protected dolaproine (27, DCC/DMAP/CSA) yielded ester 28 as a mixture of two diastereomers. After deprotection of the pyrrolidine, coupling with tripeptide 12 afforded O-silylated diazirinyl-substituted malevamide D (29, 68%). Desilylation (TBAF, THF/H2O 19:1) led to a product mixture, the separation of which required semipreparative HPLC (RP-18, MeOH/H2O 9:1). Diazirinyl-substituted malevamide 30 was obtained in 33% isolated yield as a mixture of two epimers differing at the carbinol center (HRESIMS found 863.48644, calcd. for C42H67NaF3N6O8, 863.48647). In the NMR spectra, four sets of signals were observed because of additional cis/trans isomerism of the acyl pyrrolidine moiety. As in the case of 1, the tripeptidic western half showed mostly broad signals, whereas the signals of the phenyl carbons are resolved better, even if not fully separated.
![[1860-5397-10-29-i4]](/bjoc/content/inline/1860-5397-10-29-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of photo malevamide D 30. a) NH3(l), t-BuOMe, −40 °C, 2 h, rt, 16 h, quant. b) I2 (1.2 equiv), NEt3 (2.2 equiv), Et2O, 0 °C, 15 min, rt, 1 h, 99%. c) THF, 2 N HCl, rt, 3 h, 97%. d) TBDPSCl (1.1 equiv), imidazole (2.2 equiv), DMF, 0 °C, 1 h, 80%. e) 27 (0.97 equiv), DMAP (0.41 equiv), DCC (1 equiv), CSA (0.23 equiv), 0 °C to rt, 16 h, 69%. f) TFA, DCM, 0 °C, 90 min. g) 12 (1 equiv), NEt3 (6 equiv), DEPC (2.14 equiv), DCM, 0 °C, 150 min, 68%. h) TBAF·3H2O (5 equiv), THF/H2O 19:1, 0 °C, 2 h, 33%.
Scheme 4: Synthesis of photo malevamide D 30. a) NH3(l), t-BuOMe, −40 °C, 2 h, rt, 16 h, quant. b) I2 (1.2 eq...
![[1860-5397-10-29-2]](/bjoc/content/figures/1860-5397-10-29-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: DSC curve of diazirine 25, heating rate 5 °C/min.
Figure 2: DSC curve of diazirine 25, heating rate 5 °C/min.
Cytotoxicity studies of malevamide D and diazirinyl-substituted malevamide D
The synthesized 1 proved to be potently cytotoxic with a geomean IC70 value of 1.5 nM (IC50 0.7 nM) in a panel of 42 human cancer cell lines (see Table 1 and Supporting Information File 1). The cytotoxicity was about 10-fold higher than that of paclitaxel. The breast cancer cell lines MAXF-401 (IC70 0.2 nM) and MCF-7 (0.5 nM), the colon cancer cell lines HT-29 (0.3 nM) and RKO (0.5 nM), the lung cancer cell line H460 (0.7 nM), the liver cancer cell line LIXF-575 (0.6 nM), and the renal cancer cell line RXF-393 (0.4 nM) all proved to be very sensitive. Against 36 of the 42 cell lines, IC70 values of below 10 nM were determined. The most resistant cell lines were shown to be RXF-1781 (kidney), CXF-269 (colon), MEXF-276 (melanoma, IC70 about 50 nM, each), and in particular LXFA-289 (lung, adenocarcinoma), PAXF-546L (pancreas), and PXF-1118 (pleuramesothelioma, IC70 > 100 nM, IC50 about 50 nM, each). Diazirinyl-substituted malevamide D 30 was about 200 times less cytotoxic than 1.
Table 1: Cytotoxicities (IC70, IC50, nM) of malevamide D (1) against selected human cancer cell lines.a
| histotype | cell line | IC70 | IC50 |
|---|---|---|---|
| Colon | HT-29 | 0.3 | 0.2 |
| Stomach | GXF-251 | 5.0 | 0.6 |
| Lung, adeno | LXFA-629 | 0.7 | 0.4 |
| Lung, large cell | LXFL-529 | 0.4 | 0.3 |
| Breast | MAXF-401 | 0.2 | 0.2 |
| Melanoma | MEXF-462 | 0.6 | 0.4 |
| Ovary | OVXF-899 | 1.0 | 0.7 |
| Pancreas | PAXF-1657 | 4.0 | 1.0 |
| Prostate | PRXF-22Rv1 | 0.8 | 0.6 |
| Mesothelioma | PXF-1752 | 0.4 | 0.3 |
| Kidney | RXF-486L | 0.7 | 0.5 |
| Uterus | UXF-1138L | 0.5 | 0.4 |
aFor complete results in the panel of 42 human cancer cell lines and a COMPARE analysis, see Supporting Information File 1.
To obtain clues on the likely mode of action of 1 and 30, the in vitro activity data were compared with those of 177 reference compounds by COMPARE analysis. Individual IC50 values of a test compound were correlated with those of 177 reference compounds as determined for Oncotest's 42 cell line panel and expressed quantitatively as Spearman correlation coefficients (see Supporting Information File 1). Furthermore, 1 and 30 were correlated to each other. For compound 30, COMPARE analysis revealed the highest similarities towards tubulin interacting compounds like the vinca alkaloids vinfluine, vincristine, vindesine and the taxoid compound docetaxel. Furthermore, good correlations were detected towards PLK-1 inhibitors. Similar results were obtained for 1. Determining a Spearman coefficient of 0.742 between 1 and 30 suggested that these compounds share the same mode of action, most probably antimitotic activity based on tubulin interaction.
Conclusion
Our cytotoxicity test results for 1 confirm those obtained by Scheuer and co-workers who determined IC50 values of about 0.7 nM against four cell lines (mouse lymphoma P-388, human lung carcinoma A-549, human colon carcinoma HT-29, human melanoma MEL-28) [1]. We had hoped that introduction of a Brunner-type diazirine unit in the para-position of the phenyl ring of 1 could lead to an analog that was still active, because it had been shown in several SAR studies that limited variation of the amine or alcohol component at the C-terminal amide or ester moieties of the dolastatin family members can be tolerated without much loss of cytotoxicity. This includes epimerization of the thiazole-carrying stereocenter of 3 [18], replacement of the phenyl by a methyl group, or removal of the thiazole ring [7]. Auristatins, of which some undergo clinical trials, are characterized by lacking the thiazole ring of 3 [19]. Pettit and co-workers prepared auristatin TP, which carries a phosphate unit in the para-position of the phenyl ring, and also two aminoquinoline derivatives that were as cytotoxic as dolastatin 10 [3]. Antibody conjugates of monomethylauristatin E functionalized on the C-terminal side have also shown promising pharmacological profiles [20,21]. However, compound 30 is at least 200 times less active than the natural product 1. Lower cytotoxicity may be associated with solubility problems. We will now search for more water-soluble, photoactivatable analogs of 1. Nunes and coworkers had synthesized a benzophenone-labelled derivative of the analog HTI-286, which competes with 3, and were able to identify α-tubulin as binding partner [22].
Supporting Information
| Supporting Information File 1: Procedures of synthesis and biotest, X-ray data and 1H, 13C NMR spectra of selected compounds. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Horgen, F. D.; Kazmierski, E. B.; Westenburg, H. E.; Yoshida, W. Y.; Scheuer, P. J. J. Nat. Prod. 2002, 65, 487–491. doi:10.1021/np010560r
Return to citation in text: [1] [2] [3] -
Sone, H.; Shibata, T.; Fujita, T.; Ojika, M.; Yamada, K. J. Am. Chem. Soc. 1996, 118, 1874–1880. doi:10.1021/ja9519086
Return to citation in text: [1] [2] [3] -
Pettit, G. R.; Hogan, F.; Toms, S. J. Nat. Prod. 2011, 74, 962–968. doi:10.1021/np1007334
Return to citation in text: [1] [2] [3] -
Bradley, A. M.; Devine, M.; DeRemer, D. Am. J. Health-Syst. Pharm. 2013, 70, 589–597. doi:10.2146/ajhp110608
Return to citation in text: [1] -
Pettit, G. R.; Sing, S. B.; Srirangam, J. K.; Hogan-Pierson, F.; Williams, M. D. J. Org. Chem. 1994, 59, 1796–1800. doi:10.1021/jo00086a034
Return to citation in text: [1] -
Reddy, G. V.; Rao, G. V.; Sreevani, V.; Iyngar, D. S. Tetrahedron Lett. 2000, 41, 949–951. doi:10.1016/S0040-4039(99)02105-X
Return to citation in text: [1] -
Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Chem. Pharm. Bull. 1995, 43, 1706–1718. doi:10.1248/cpb.43.1706
Return to citation in text: [1] [2] -
Maugras, I.; Poncet, J.; Jouin, P. Tetrahedron 1990, 46, 2807–2816. doi:10.1016/S0040-4020(01)88373-3
Return to citation in text: [1] -
Cella, R.; Venturoso, R. C.; Stefani, H. A. Tetrahedron Lett. 2008, 49, 16–19. doi:10.1016/j.tetlet.2007.11.031
Return to citation in text: [1] -
Bowman, R. E.; Stroud, H. H. J. Chem. Soc. 1950, 1342–1345. doi:10.1039/jr9500001342
Return to citation in text: [1] -
Aurelio, L.; Box, J. S.; Brownlee, R. T. C.; Hughes, A. B.; Sleebs, M. M. J. Org. Chem. 2003, 68, 2652–2667. doi:10.1021/jo026722l
Return to citation in text: [1] -
Benedetti, E.; Carlomagno, T.; Fraternali, F.; Hamada, Y.; Hayashi, K.; Paolillo, L.; Shioiri, T. Biopolymers 1995, 36, 525–538. doi:10.1002/bip.360360414
Return to citation in text: [1] -
Harrigan, G. G.; Luesch, H.; Yoshida, W. Y.; Moore, R. E.; Nagle, D. G.; Paul, V. J.; Mooberry, S. L.; Corbett, T. H.; Valeriote, F. A. J. Nat. Prod. 1998, 61, 1075–1077. doi:10.1021/np980321c
Return to citation in text: [1] -
Boyd, V. L.; Bozzini, M.; Guga, P. J.; DeFranco, R. J.; Yuan, P.-M.; Loudon, G. M.; Nguyen, D. J. Org. Chem. 1995, 60, 2581–2587. doi:10.1021/jo00113a042
Return to citation in text: [1] -
Wartmann, T.; Lindel, T. Eur. J. Org. Chem. 2013, 1649–1652. doi:10.1002/ejoc.201201726
Return to citation in text: [1] -
Bolli, M.; Lehmann, D.; Mathys, B.; Müller, C.; Nayler, O.; Velker, J.; Weller, T. Novel thiophene derivatives. WO Patent WO 2006/100635 A2, Sept 28, 2006.
Return to citation in text: [1] -
CCDC 956232 contains the supplementary crystallographic data of compound (E)-22, which can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (12, Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or deposit@ccdc.cam.ac.uk).
Return to citation in text: [1] -
Bai, R.; Roach, M. C.; Jayaram, S. K.; Barkoczy, J.; Pettit, G. R.; Ludueña, R. F.; Hamel, E. Biochem. Pharmacol. 1993, 45, 1503–1515. doi:10.1016/0006-2952(93)90051-W
Return to citation in text: [1] -
Shnyder, S. D.; Cooper, P. A.; Millington, N. J.; Pettit, G. R.; Bibby, M. C. Int. J. Oncol. 2007, 31, 353–360.
Return to citation in text: [1] -
Doronina, S. O.; Bovee, T. D.; Meyer, D. W.; Miyamoto, J. B.; Anderson, M. E.; Morris-Tilden, C. A.; Senter, P. D. Bioconjugate Chem. 2008, 19, 1960–1963. doi:10.1021/bc800289a
Return to citation in text: [1] -
Axup, J. Y.; Bajjuri, K. M.; Ritland, M.; Hutchins, B. M.; Kim, C. H.; Kazane, S. A.; Halder, R.; Forsyth, J. S.; Santidrian, A. F.; Stafin, K.; Lu, Y.; Tran, H.; Seller, A. J.; Biroc, S. L.; Szydlik, A.; Pinkstaff, J. K.; Tian, F.; Sinha, S. C.; Felding-Habermann, B.; Smider, V. V.; Schultz, P. G. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16101–16106. doi:10.1073/pnas.1211023109
Return to citation in text: [1] -
Nunes, M.; Kaplan, J.; Wooters, J.; Hari, M.; Minnick, A. A., Jr.; May, M. K.; Shi, C.; Musto, S.; Beyer, C.; Krishnamurthy, G.; Qiu, Y.; Loganzo, F.; Ayral-Kaloustian, S.; Zask, A.; Greenberger, L. M. Biochemistry 2005, 44, 6844–6857. doi:10.1021/bi0474766
Return to citation in text: [1]
| 1. | Horgen, F. D.; Kazmierski, E. B.; Westenburg, H. E.; Yoshida, W. Y.; Scheuer, P. J. J. Nat. Prod. 2002, 65, 487–491. doi:10.1021/np010560r |
| 2. | Sone, H.; Shibata, T.; Fujita, T.; Ojika, M.; Yamada, K. J. Am. Chem. Soc. 1996, 118, 1874–1880. doi:10.1021/ja9519086 |
| 14. | Boyd, V. L.; Bozzini, M.; Guga, P. J.; DeFranco, R. J.; Yuan, P.-M.; Loudon, G. M.; Nguyen, D. J. Org. Chem. 1995, 60, 2581–2587. doi:10.1021/jo00113a042 |
| 4. | Bradley, A. M.; Devine, M.; DeRemer, D. Am. J. Health-Syst. Pharm. 2013, 70, 589–597. doi:10.2146/ajhp110608 |
| 3. | Pettit, G. R.; Hogan, F.; Toms, S. J. Nat. Prod. 2011, 74, 962–968. doi:10.1021/np1007334 |
| 3. | Pettit, G. R.; Hogan, F.; Toms, S. J. Nat. Prod. 2011, 74, 962–968. doi:10.1021/np1007334 |
| 12. | Benedetti, E.; Carlomagno, T.; Fraternali, F.; Hamada, Y.; Hayashi, K.; Paolillo, L.; Shioiri, T. Biopolymers 1995, 36, 525–538. doi:10.1002/bip.360360414 |
| 2. | Sone, H.; Shibata, T.; Fujita, T.; Ojika, M.; Yamada, K. J. Am. Chem. Soc. 1996, 118, 1874–1880. doi:10.1021/ja9519086 |
| 13. | Harrigan, G. G.; Luesch, H.; Yoshida, W. Y.; Moore, R. E.; Nagle, D. G.; Paul, V. J.; Mooberry, S. L.; Corbett, T. H.; Valeriote, F. A. J. Nat. Prod. 1998, 61, 1075–1077. doi:10.1021/np980321c |
| 8. | Maugras, I.; Poncet, J.; Jouin, P. Tetrahedron 1990, 46, 2807–2816. doi:10.1016/S0040-4020(01)88373-3 |
| 9. | Cella, R.; Venturoso, R. C.; Stefani, H. A. Tetrahedron Lett. 2008, 49, 16–19. doi:10.1016/j.tetlet.2007.11.031 |
| 2. | Sone, H.; Shibata, T.; Fujita, T.; Ojika, M.; Yamada, K. J. Am. Chem. Soc. 1996, 118, 1874–1880. doi:10.1021/ja9519086 |
| 7. | Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Chem. Pharm. Bull. 1995, 43, 1706–1718. doi:10.1248/cpb.43.1706 |
| 1. | Horgen, F. D.; Kazmierski, E. B.; Westenburg, H. E.; Yoshida, W. Y.; Scheuer, P. J. J. Nat. Prod. 2002, 65, 487–491. doi:10.1021/np010560r |
| 6. | Reddy, G. V.; Rao, G. V.; Sreevani, V.; Iyngar, D. S. Tetrahedron Lett. 2000, 41, 949–951. doi:10.1016/S0040-4039(99)02105-X |
| 5. | Pettit, G. R.; Sing, S. B.; Srirangam, J. K.; Hogan-Pierson, F.; Williams, M. D. J. Org. Chem. 1994, 59, 1796–1800. doi:10.1021/jo00086a034 |
| 10. | Bowman, R. E.; Stroud, H. H. J. Chem. Soc. 1950, 1342–1345. doi:10.1039/jr9500001342 |
| 11. | Aurelio, L.; Box, J. S.; Brownlee, R. T. C.; Hughes, A. B.; Sleebs, M. M. J. Org. Chem. 2003, 68, 2652–2667. doi:10.1021/jo026722l |
| 17. | CCDC 956232 contains the supplementary crystallographic data of compound (E)-22, which can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (12, Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or deposit@ccdc.cam.ac.uk). |
| 15. | Wartmann, T.; Lindel, T. Eur. J. Org. Chem. 2013, 1649–1652. doi:10.1002/ejoc.201201726 |
| 16. | Bolli, M.; Lehmann, D.; Mathys, B.; Müller, C.; Nayler, O.; Velker, J.; Weller, T. Novel thiophene derivatives. WO Patent WO 2006/100635 A2, Sept 28, 2006. |
| 22. | Nunes, M.; Kaplan, J.; Wooters, J.; Hari, M.; Minnick, A. A., Jr.; May, M. K.; Shi, C.; Musto, S.; Beyer, C.; Krishnamurthy, G.; Qiu, Y.; Loganzo, F.; Ayral-Kaloustian, S.; Zask, A.; Greenberger, L. M. Biochemistry 2005, 44, 6844–6857. doi:10.1021/bi0474766 |
| 3. | Pettit, G. R.; Hogan, F.; Toms, S. J. Nat. Prod. 2011, 74, 962–968. doi:10.1021/np1007334 |
| 20. | Doronina, S. O.; Bovee, T. D.; Meyer, D. W.; Miyamoto, J. B.; Anderson, M. E.; Morris-Tilden, C. A.; Senter, P. D. Bioconjugate Chem. 2008, 19, 1960–1963. doi:10.1021/bc800289a |
| 21. | Axup, J. Y.; Bajjuri, K. M.; Ritland, M.; Hutchins, B. M.; Kim, C. H.; Kazane, S. A.; Halder, R.; Forsyth, J. S.; Santidrian, A. F.; Stafin, K.; Lu, Y.; Tran, H.; Seller, A. J.; Biroc, S. L.; Szydlik, A.; Pinkstaff, J. K.; Tian, F.; Sinha, S. C.; Felding-Habermann, B.; Smider, V. V.; Schultz, P. G. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16101–16106. doi:10.1073/pnas.1211023109 |
| 7. | Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Chem. Pharm. Bull. 1995, 43, 1706–1718. doi:10.1248/cpb.43.1706 |
| 19. | Shnyder, S. D.; Cooper, P. A.; Millington, N. J.; Pettit, G. R.; Bibby, M. C. Int. J. Oncol. 2007, 31, 353–360. |
| 1. | Horgen, F. D.; Kazmierski, E. B.; Westenburg, H. E.; Yoshida, W. Y.; Scheuer, P. J. J. Nat. Prod. 2002, 65, 487–491. doi:10.1021/np010560r |
| 18. | Bai, R.; Roach, M. C.; Jayaram, S. K.; Barkoczy, J.; Pettit, G. R.; Ludueña, R. F.; Hamel, E. Biochem. Pharmacol. 1993, 45, 1503–1515. doi:10.1016/0006-2952(93)90051-W |
© 2014 Telle et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)