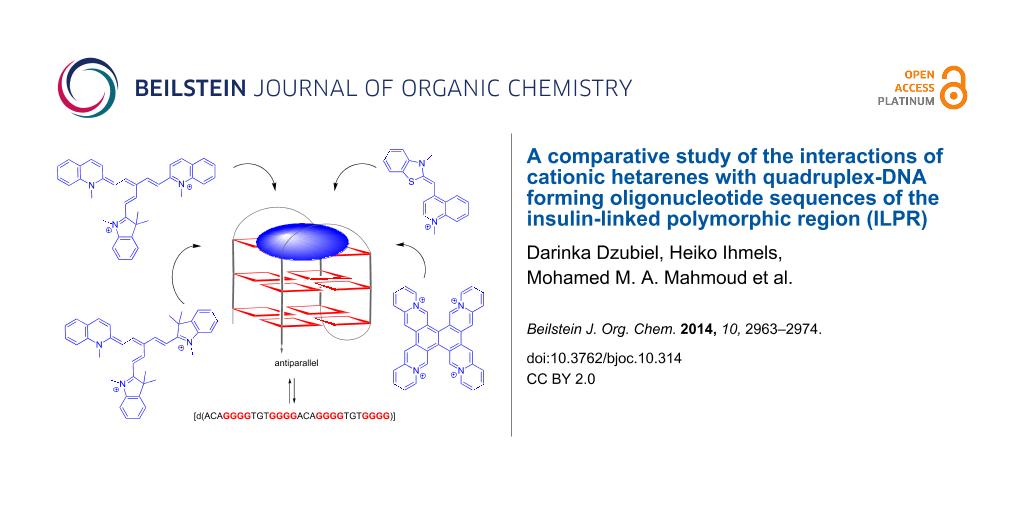
Abstract
The interactions of the ILPR sequence (ILPR = "insulin-linked polymorphic region") a2 [d(ACAG4TGTG4ACAG4TGTG4)] with [2.2.2]heptamethinecyanine derivatives 1a–e and with the already established quadruplex ligands coralyne (2), 3,3′-[2,6-pyridinediylbis(carbonylimino)]bis[1-methylquinolinium] (3), 4,4′,4′′,4′′′-(21H,23H-porphine-5,10,15,20-tetrayl)tetrakis[1-methylpyridinium] (4), naphtho[2,1-b:3,4-b′:6,5-b′′:7,8-b′′′]tetraquinolizinium (5) and thiazole orange (6) were studied. It is demonstrated with absorption, fluorescence and CD spectroscopy that all investigated ligands bind with relatively high affinity to the ILPR-quadruplex DNA a2 (0.2–5.5 × 106 M−1) and that in most cases the binding parameters of ligand-ILPR complexes are different from the ones observed with other native quadruplex-forming DNA sequences.

Graphical Abstract
Introduction
The “insulin-linked polymorphic region” (ILPR) is a physiologically relevant G-rich DNA sequence that consists of repetitive DNA units with varying length and sequence [1]. The ILPR is located in the promoter region of the human insulin gene and is proposed to control the expression of the latter. It was shown that insulin-dependent diabetes mellitus (IDDM, type-I diabetes) is associated with the number of these minisatellites. Specifically, the number of these repeating units varies from ca. 160 in healthy humans to ca. 40 in IDDM patients [2]. From the 14 known repeating units of ILPR the variants a [d(ACAGGGGTGTGGGG)], b [d(ACAGGGGTCTGGGG)] and c [d(ACAGGGGTCCTGGGG)] occur most frequently; and a has the highest transcriptional activity [2,3]. It was shown that the sequences a–c form stable G-quadruplex structures in vitro and that this tendency is even enhanced by binding of the insulin protein [4-8]. In addition, it was demonstrated that the nucleotide sequence a2, [d(ACAG4TGTG4ACAG4TGTG4)], has parallel and antiparallel quadruplex forms that coexist under physiological conditions (Scheme 1) [9]. Notably, the biological relevance of ILPR quadruplexes has been suggested considering recent discoveries on the influence of quadruplex formation on the function of nucleic acids and the observation that insulin associates with quadruplex DNA [10]. In particular, it was assumed that the formation of quadruplex structures affects the transcriptional activity, because the position of the ILPR in the promoter region of the gene represents a stake close to the transcription process [9,11,12]. Furthermore, quadruplex structures may interfere with the replication process because of their thermal and mechanical stability. Namely, the force required to unfold the quadruplex is larger than the one to block helicases [9,13], such that quadruplex structures reduce the activity of these enzymes to greater extent than the corresponding duplex DNA [14]. There is also evidence that longer ILPR sequences may form multiple quadruplex structures that interact with each other, and it was speculated that the observed increase in replication errors in minisatellite regions is related with these higher-order DNA structures [15].
![[1860-5397-10-314-i1]](/bjoc/content/inline/1860-5397-10-314-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Equilibrium between single-stranded ILPR-DNA a2 and its parallel and antiparallel quadruplex form.
Scheme 1: Equilibrium between single-stranded ILPR-DNA a2 and its parallel and antiparallel quadruplex form.
It has been demonstrated throughout the last decade that quadruplex DNA-binding ligands have a large potential to increase its stability towards unfolding and to influence the equilibrium between different quadruplex forms by stabilizing one particular quadruplex conformation [16,17]. Therefore, it is proposed that such ligands may interfere with physiological processes that involve quadruplex DNA. In this context, it is remarkable that systematic studies on the interaction of ligands with ILPR-DNA are rather rare. To the best of our knowledge there exists one study to develop a fluorescence screening method for the identification of selective quadruplex ligands in which, along with other sequences, an ILPR sequence has been used [18,19]. To fill this gap of knowledge, we investigated the interactions of a series of quadruplex ligands with the representative ILPR sequence a2. As we have demonstrated already that the [2.2.2]heptamethinecyanine dye binds selectively to quadruplex DNA and that its interaction with the quadruplex is indicated by a drastic emission light-up effect [20,21], we chose the [2.2.2]heptamethinecyanine derivatives 1a–e [22] as ligands for this study. For a better comparison of data we also included the already established quadruplex ligands 2–6 [23-27] in this study (Figure 1).
![[1860-5397-10-314-1]](/bjoc/content/figures/1860-5397-10-314-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of ligands 1–6 used in this study.
Figure 1: Structures of ligands 1–6 used in this study.
Results
Thermal DNA-denaturation experiments
The temperature-dependent structural changes of ILPR-DNA were analyzed by CD-spectroscopic and fluorimetric analysis. CD spectra were obtained from a solution of the ILPR quadruplex-forming oligonucleotide a2 in K+-containing buffer (95 mM) in a temperature range from 20 to 95 °C. The oligonucleotide a2 showed CD signals that are characteristic of a mixture of parallel (λmax = 265 nm; λmin = 235 nm) and antiparallel (λmax = 295 nm) quadruplex structures (Figure 2A) [9,28]. With increasing temperature, the intensity of the maxima at 210 and 265 nm as well as the ones of the minimum at 235 nm decrease at T > 60 °C. On the other hand, the intensity of the signal at 295 nm slightly increases at T > 60 °C and then decreases at T > 80 °C (Figure 1A and 1B). These results are in agreement with literature data [9]; however, in the latter case the signal at 265 nm increased at 60 °C < T > 80 °C instead of the signal at 290 nm, presumably because different buffer solutions were used in the latter study. It should be noted that the CD spectrum of a2 in potassium phosphate buffer (95 mM K+) changes slightly with time. Specifically, the intensity of the CD bands at 210, 235 and 265 nm decreases slowly within 48 h, whereas the maximum at 295 nm remains essentially the same in this time range (Figure 3).
![[1860-5397-10-314-2]](/bjoc/content/figures/1860-5397-10-314-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A) CD spectra of ILPR-DNA a2 (20 μM) at different temperatures in potassium phosphate buffer (95 mM, pH 7.0). Arrows indicate the development of the bands with increasing temperature. B) Plot of the intensity changes (CD/CD0) of the CD signals at 265 nm (■) and 295 nm (○) versus temperature.
Figure 2: A) CD spectra of ILPR-DNA a2 (20 μM) at different temperatures in potassium phosphate buffer (95 mM...
![[1860-5397-10-314-3]](/bjoc/content/figures/1860-5397-10-314-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CD spectra of ILPR-sequence a2 (20 µM) in potassium phosphate buffer (95 mM, pH 7.0; __ : immediately after the preparation of the DNA stock solution; ···· : 2 d after the preparation).
Figure 3: CD spectra of ILPR-sequence a2 (20 µM) in potassium phosphate buffer (95 mM, pH 7.0; __ : immediate...
The thermal stability of the ILPR-DNA with respect to unfolding was also examined by emission spectroscopy in K+- and Na+-containing buffer, i.e., the melting temperature, Tm, of the dye-labelled quadruplex-forming ILPR sequence Fa2T [fluorescein-d(ACAG4TGTG4ACAG4TGTG4)-tetramethylrhodamine] was determined by fluorimetric monitoring of the temperature-dependent Förster resonance energy transfer (FRET) between the dyes [29]. In sodium cacodylate buffer (10 mM Na+, 10 mM K+, 90 mM Li+) the melting curve of the DNA has a weak transition at 50 °C and a more pronounced one at Tm = 71.0 °C (Figure 4). The results of the melting experiments always refer to the latter transition. In potassium phosphate buffer (95 mM K+) the melting temperature of the DNA is Tm = 87.8 °C. This temperature is relatively high, so that further stabilization by a ligand leads to a Tm close to the boiling point of the solvent. Therefore, studies of ligand–DNA complexes were not performed in this medium.
![[1860-5397-10-314-4]](/bjoc/content/figures/1860-5397-10-314-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorimetric monitoring of thermal DNA-denaturation of the ILPR quadruplex Fa2T (0.2 μM DNA concentration in oligonucleotide) in potassium phosphate buffer (····, 95 mM, pH 7.0) and KCl-LiCl-Na-cacodylate buffer (—, 10 mM K+, 90 mM Li+, pH 7.2). Inset: Plot of the first derivative f' (T) of the melting curves.
Figure 4: Fluorimetric monitoring of thermal DNA-denaturation of the ILPR quadruplex Fa2T (0.2 μM DNA concent...
The influence of the ligands 1–6 on the melting temperature of ILPR-DNA was determined by fluorimetric monitoring of the DNA melting curves of Fa2T at different ligand–DNA ratios, LDR (Table 1, Figures S1 and S2 in Supporting Information File 1). Most of the tested ligands, i.e. 1a, 1c, 1d, 2, 4, 5, and 6, induced only a moderate increase of the melting temperature of the ILPR-quadruplex (ΔTm = ca. 2–6 °C). Nevertheless, the addition of the cyanine derivative 1b or the bis-quinolinium derivative 3 induced a significantly larger shift of the quadruplex melting temperature (1b: ΔTm = 14.5 °C; 3: 14.9 °C; LDR = 5.0).
Table 1: Shifts of melting temperature, ΔTm, of quadruplex-DNA Fa2T induced by ligands 1–6 as determined from fluorimetric thermal DNA denaturation studies.
| LDR | ΔTm [°C] a | ||
|---|---|---|---|
| 1.3 | 2.5 | 5.0 | |
| 1a | 1.0 | 1.9 | 4.9 |
| 1b | 0.8 | 7.1 | 14.5 |
| 1c | 0.1 | 1.1 | 3.5 |
| 1d | 0.5 | 4.0 | 3.7 |
| 1e | 1.3 | 2.2 | 3.4 |
| 2 | 0.7 | 1.7 | 3.1 |
| 3 | 3.9 | 6.4 | 14.9 |
| 4 | 1.4 | 2.1 | 6.3 |
| 5 | 0.1 | 1.5 | 5.8 |
| 6 | 0.7 | 0.8 | 1.8 |
aΔTm of Fa2T; cDNA = 0.2 µM (in oligonucleotide); KCl-LiCl-Na-cacodylate buffer (10 mM K+, 90 mM Li+, pH 7.2); λex = 470 nm; λem = 515 nm; estimated error: ±0.5 °C of the given data.
Photometric and fluorimetric titrations
The interactions of the ligands 1–6 with ILPR-DNA a2 were further analyzed with photometric and fluorimetric titrations (Figure 5 and Figure 6, Table 2). In general, a hypochromic effect and a bathochromic shift of the absorption maximum of the ligand were observed upon addition of DNA (Supporting Information File 1, Figures S3 and S5). In the case of coralyne (2) and the tetraazoniahetarene derivative 5 this effect is only weakly pronounced. Only during the titration of the bis-quinolinium derivative 3 an isosbestic point developed at 364 nm.
![[1860-5397-10-314-5]](/bjoc/content/figures/1860-5397-10-314-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fluorimetric titration of 1a (A), 1b (B), 1c (C), 1d (D) and 1e (E) with a2 in potassium phosphate buffer (95 mM, pH 7.0); A, B, E: cLig = 5.0 μM; C, D: cLig = 10 μM; A: λex = 385 nm; B: λex = 364 nm; C: λex = 431 nm; D, E: λex = 580 nm. Arrows indicate the development of the bands with increasing DNA concentration. Inset: Plot of the normalized emission intensity versus DNA concentration. Lines denote the best fit of experimental data to the theoretical model.
Figure 5: Fluorimetric titration of 1a (A), 1b (B), 1c (C), 1d (D) and 1e (E) with a2 in potassium phosphate ...
![[1860-5397-10-314-6]](/bjoc/content/figures/1860-5397-10-314-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fluorimetric titration of 2 (A), 3 (B), 4 (C), 5 (D) and 6 (E) with a2 in potassium phosphate buffer (95 mM, pH 7.0); A, B, D, E: cLig = 5.0 μM; C: cLig = 10 μM; A, D: λex = 385 nm; B: λex = 364 nm; C: λex = 431 nm; E: λex = 490 nm. Inset: Plot of the change of the emission intensity versus DNA concentration. Lines denote the best fit of experimental data to the theoretical model. Arrows indicate the development of the bands with increasing DNA concentration.
Figure 6: Fluorimetric titration of 2 (A), 3 (B), 4 (C), 5 (D) and 6 (E) with a2 in potassium phosphate buffe...
Table 2: Absorption and emission properties of ligands 1–6 in the absence and presence of ILPR-DNA a2, and binding constants Kb.
| Ligand | λAbs [nm]a | λem [nm]b | I/I0 c | Kba2 [106 M−1]d | Kb22AG [106 M−1]e | |
|---|---|---|---|---|---|---|
| free | bound | |||||
| 1a | 624 | 635 | 712 | 92 | 1.2 | 0.8f |
| 1b | 577 | 648 | 700 | 17 | 2.7 | 0.2 |
| 1c | 635 | 740 | 769 | 18 | 0.2 | 0.2 |
| 1d | 626 | 648 | 718 | 128 | 5.5 | 0.9 |
| 1e | 628 | 640 | 703 | 87 | 17 | 0.4 |
| 2 | 295 | 435 | 480 | <0.1 | 2.5 | n.d.g |
| 3 | 346 | 374 | 404 | <0.1 | 2.4 | n.d.g |
| 4 | 422 | 440 | 665, 728 | n.d.g | 2.2 | 2.9h |
| 5 | 387 | 395 | 513 | <0.1 | 3.5 | 1.8h |
| 6 | 473 | 513 | 530 | 1766 | 0.3 | 2.1i |
aLong-wavelength absorption maximum of the unbound and quadruplex-bound ligand. bEmission maximum of bound ligand. cRelative emission intensity, I/I0 (I = emission intensity of DNA-bound ligand at saturation, I0 = emission of unbound ligand). dBinding constant of ligand–a2 complex, Kba2, determined from fluorimetric titrations; DNA concentration in nucleotide bases. eBinding constant of ligand–22AG complex, Kb22AG, determined either from fluorimetric titrations or literature data; DNA concentration in oligonucleotide. fRef. [20]. gn.d. = not determined. hRef. [23]. iRef [30].
The addition of ILPR-DNA a2 to the cyanine derivatives 1a–e and 6 resulted in a strong increase of the emission intensity (light-up effect) with increasing concentration of a2 (Figure 5 and Figure 6E). This effect was most pronounced in the case of thiazole orange (6) whose emission intensity increases by a factor of I/I0 = 1766. Within the series of heptamethine cyanine dyes 1a–e, the derivative 1d exhibits the largest light-up factor with I/I0 = 128. In contrast, the emission of coralyne (2), bis-quinolinium derivative 3 and the tetraazoniahetarene derivative 5 was quenched upon addition of a2 (Figure 6A, 6B, and 6D; Table 2). The data of the fluorimetric titrations was used to estimate the binding constants Kba2 from a fit of the experimentally determined binding isotherms to the theoretical model [31] with resulting binding constants in the range of 0.2–5.5 × 106 M−1 (Table 2). Notably, the development of the emission intensity of ligands 1d and 1e showed two distinctly different trends during the titration with a2, which indicates two different binding modes (Supporting Information File 1, Figure S4). Specifically, at LDR values <1.1 (1d) or <0.8 (1e) the emission intensity is significantly less quenched as compared to the beginning of the titration. The binding constants of the ligand–DNA complexes formed at the beginning of the titrations were estimated assuming that the emission intensities of 1d und 1e at LDR = 1.1 and 0.8 essentially represent the one of the bound ligand in this particular binding mode. Hence, the binding constants were determined from the respective binding isotherms (inset in Figure 5D and 5E; 1d: Kba2 = 5.5 × 106 M−1; 1e: Kba2 = 1.7 × 107 M−1). For comparison, fluorimetric titrations of the telomeric quadruplex 22AG [5'-A(GGGTTA)3GGG-3'] to the cyanine derivatives 1b–e were performed and the binding constants were obtained from the analysis of these data (Table 2; Figure S6 in Supporting Information File 1).
The stoichiometry of the complexes of the ILPR-DNA a2 with the representative ligands 1e, 4 and 6 was determined with the continuous variations method (Job plot analysis). For that purpose, the emission intensity was plotted versus the mole fraction of the ligand, XLigand, at constant total concentration of ligand and DNA (Figure 7). The graphically obtained maxima, i.e., the intercept of linearly fitted ascending and descending curve segments, were located at 0.48, 0.73 and 0.50 for ligands 1e, 4 and 6, respectively. Within the error margin these data correspond to a binding stoichiometry of 1:1 for complexes of 1e and 6 with DNA a2, and a stoichiometry of 3:1 for the assembly of the porphyrin derivative 4 with the ILPR-DNA. However, it should be noted that strong fluctuations in the Job plot of the complex formation between ligand 1e and ILPR-DNA a2, especially between XLigand = 0–0.4, indicate strong heterogeneous binding under these conditions.
![[1860-5397-10-314-7]](/bjoc/content/figures/1860-5397-10-314-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Job plot from fluorimetric analysis of mixtures of ligands 1e (A), 4 (B) and 6 (C) with ILPR-DNA a2 (cLigand + cDNA = 10.0 μM) in potassium phosphate buffer (95 mM, pH 7.0); XLigand = mole fraction of the ligand; A: λex = 580 nm; B: λex = 431 nm; C: λex = 490 nm; T = 20 °C.
Figure 7: Job plot from fluorimetric analysis of mixtures of ligands 1e (A), 4 (B) and 6 (C) with ILPR-DNA a2...
CD spectroscopic analysis of ligand–DNA interactions
The interactions of the ligands 1d, 1e, 2, 4–6 with ILPR-DNA a2 were analyzed by CD spectroscopy (Figure 8, Figure S7 in Supporting Information File 1). In most cases, the weak CD signal at 265 nm decreases and the CD signal at 295 nm increases slightly when the ligands were added. Upon addition of the cyanine derivative 1e to ILPR-DNA, the intensity of the peak at 295 nm increased, whereas the peak at 265 nm slightly decreased. On the other hand, only very small, insignificant changes were observed on titration of 1d to ILPR-DNA (Figure 8, Figure S7 in Supporting Information File 1). Notably, the addition of the porphyrin 4 and tetraazoniahetarene 5 to ILPR-DNA a2 has a strong effect on the positive CD signal at 265 nm, leading to the development of a negative band at 260 nm at high LDR ratio (Figure 8D and 8E). In addition, induced CD (ICD) signals were observed only in the absorption range of compounds 4–6.
![[1860-5397-10-314-8]](/bjoc/content/figures/1860-5397-10-314-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: CD spectra of ILPR-DNA a2 in the presence of 1d (A), 1e (B), 2 (C), 4 (D), 5 (E) and 6 (F) at LDR = 0 (black), 0.5 (red), 1 (blue), 2 (magenta); cDNA = 20 μM in potassium phosphate buffer (95 mM, pH 7.0); T = 20 °C. Inset: Magnified ICD signals of the bound ligands (magnification factor: ca. 10).
Figure 8: CD spectra of ILPR-DNA a2 in the presence of 1d (A), 1e (B), 2 (C), 4 (D), 5 (E) and 6 (F) at LDR =...
Discussion
Generally, it is possible to distinguish between different G-quadruplex DNA forms with the aid of CD spectroscopy [32]. Hence, in agreement with literature data [9,10], the characteristic bands at 235, 265 and 295 nm (Figure 2) show that the employed ILPR-DNA a2 exists as a mixture of parallel (265 nm) and antiparallel (295 nm) G-quadruplex structures in potassium phosphate buffer solution. The presence of different quadruplex forms is further supported by the temperature-dependent CD spectra that denote the different melting temperatures of the parallel and antiparallel forms (Figure 3). Although it has been reported that the population of the parallel form increases with a low concentration of K+ ions [9]; in our hands it turned out to be dependent also on handling procedure. Hence, if the ILPR-quadruplex a2 is heated to 97 °C and then cooled immediately to room temperature within 30 s, the CD spectrum of the solution shows a stronger CD signal at 265 nm than at 295 nm [9]. In contrast, if the ILPR-quadruplex a2 is cooled slowly from 97 °C to room temperature with a rate of 0.5 °C/min the signal at 295 nm is higher than the one at 265 nm [10]. These results clearly demonstrate a delicate equilibrium between the different quadruplex forms and some kinetic barriers that result in rather slow interconversion processes. Nevertheless, to provide comparable data, in this study we used slowly cooled ILPR-quadruplex a2 to ensure that we have the same relative population of parallel and antiparallel forms in each experiment.
The fluorimetric thermal DNA denaturation experiments show that the ligands 1b and 3 induce the highest stabilization of ILPR-quadruplex under the employed conditions (ΔTm = 14.5 °C, 14.9 °C respectively, Table 1), whereas the ligands 1a, 1c–e, 2, 4 and 5 stabilize this DNA just moderately (ΔTm = ca. 3–6 °C). Although the cyanine derivatives 1d and 1e show the highest binding affinity towards ILPR-quadruplex, these ligands induce only a moderate shift of its melting temperature (Table 2). This apparent contradiction may be the result of different buffer solutions used in different types of experiments, because the buffer composition, especially the K+ concentration, affects the population of parallel and antiparallel forms [9]. In the thermal denaturation experiment, the population of the parallel forms increases, because a low K+ concentration buffer was used (vide supra). On the other hand, a larger concentration of K+ was used during the fluorimetric titration leading to a higher fraction of the antiparallel form [9,10]. Apart from the derivatives 1c and 6 the binding constants of the cyanine dyes with the ILPR-DNA are larger than those observed with the telomeric quadruplex 22AG (Table 2). For example, the cyanine derivative 1e shows the most significant difference between the binding affinity towards ILPR and the telomeric quadruplex (a2: Kba2 = 1.7 × 107 M−1; 22AG: Kb22AG = 3.9 × 105 M−1). Although thiazole orange (6) has a relatively high binding constant with telomeric quadruplex DNA, it has a lower affinity towards the ILPR quadruplex under the same conditions. In addition, thiazole orange (6) shows the least pronounced stabilization effect in the thermal denaturation experiment (Table 2). As cyanine dyes bind to quadruplex DNA by terminal π-stacking [20], which is in agreement with the observed binding stoichiometry of 1:1 (Figure 7), the different composition of the loops in ILPR-quadruplex (ACA and TGT in ILPR versus TTA in telomeric quadruplex) may affect the binding affinity of ligands towards the ILPR-DNA. On the other hand, the porphyrin 4 and the tetraazoniahetarene derivative 5 show similar binding affinities towards the ILPR-quadruplex thus resembling their interactions with the telomeric 22AG quadruplex [23]. This observation indicates that these ligands may bind in similar types of binding sites. It should be noted that in the case of the porphyrin 4 the ligand–a2 stoichiometry of 1:3 (Figure 7) indicates additional stacking of the ligand along the phosphate backbone of the ILPR-DNA.
The cyanine derivatives 1a, 1d, 1e and 6 show significant light-up effects upon association with the ILPR quadruplex (Table 2). Although thiazole orange (6) shows the highest light-up factor (I/I0 = 1766), it has a low selectivity over different types of DNA [33]. Thus, the use of thiazole orange for the selective fluorimetric detection of quadruplex DNA is rather limited. On the other hand, the cyanine derivatives 1a, 1d and 1e show high selectivity towards quadruplex as compared to double stranded DNA [20]. Therefore, these ligands may be used as probes for the detection of quadruplex DNA.
Although the CD-spectroscopic analysis only provides qualitative information about the binding between the ligand and the DNA, they may be used to identify some general trends. Thus, the CD-spectroscopic analysis revealed that some of the ligands induce a very small (1d, 6), moderate (2), or even strong (1e) increase of the CD band of the DNA a2 at 295 nm. As this particular band has been assigned to the antiparallel quadruplex structure of a2 [9,10] the induced increase may indicate a stabilization of this DNA structure upon complex formation. Moreover, a comparison with the telomeric quadruplex 22AG shows that the ligands 1d, 1e and 6 induce the same effect on the CD band of this quadruplex at 295 nm (Supporting Information File 1, Figure S8). At the same time, the ICD of thiazole orange (6) in the presence of 22AG is stronger than that with a2 (Figure 8F and Figure S8C, Supporting Information File 1) indicating a stronger interaction of the ligand towards the telomeric quadruplex. This observation is in agreement with the higher affinity of 6 towards 22AG (Table 2). Notably, coralyne (2) has a significantly different influence on the CD spectrum of 22AG as compared to a2. In contrast to ILPR-DNA a2, the band of 22AG at 295 nm decreases upon addition of compound 2, along with a strong ICD signal of the ligand [34]; thus indicating a different binding mode of coralyne (2) with a2 and 22AG.
Notably, the CD-spectroscopic analysis shows a marginal effect of 1d and 1e on the ILPR-DNA a2. Nevertheless, as the other experimental data confirm the association of this ligand to the quadruplex it may be concluded that the complex formation between a2 and 1d and 1e does not lead to a significant change of the DNA structure.
In the case of ligands 2, 4, 5 and 6, a negative CD band developed at 265 nm together with a weak positive band with a maximum at ca. 240 nm upon association with quadruplex a2 (Figure 8). Along with the increase of the CD signal at 295 nm these developments may indicate the pronounced formation of an antiparallel quadruplex form [35]. Furthermore, the CD spectrum of a2 in the presence of the ligands 2, 4, 5 and 6 displays roughly the same maxima as the antiparallel quadruplex with only two stacked G-quartet planes that is formed by the thrombin binding aptamer (TBA) [d(G2T2G2TGTG2T2G2)], though with varying intensities and slightly different shifts [32]. In addition, it has been suggested that the porphyrin 4 also binds to an antiparallel quadruplex form that consists of just two stacked G-quartets and more extended loop structures [36], the so-called Form 3. Presumably, the sterically demanding ligand 4 fits well into the larger binding sites of the Form 3 quadruplex [37,38]. Considering all these observations it may be very carefully proposed that the ILPR-DNA a2 also forms structures with only two stacked G-quartets that are stabilized upon association of the ligands 2, 4, 5 and 6. Nevertheless, this proposed folding pattern of the oligonucleotide a2 needs to be confirmed in a more detailed structure analysis by high-resolution NMR spectroscopy.
Conclusion
This study represents the first comparative investigation of the interaction of different quadruplex ligands with an oligonucleotide sequence from the ILPR. In summary, it is demonstrated that in most cases the binding parameters of ligand-ILPR complexes are different from the ones observed with other native quadruplex-forming DNA sequences. All ligands under investigation show a high binding affinity towards the ILPR quadruplex. Cyanine derivatives – except thiazole orange (6) – have a higher affinity towards ILPR quadruplex than towards the telomeric quadruplex. From these results it may be concluded that, in principle, the association of exogenous ligands with the ILPR-DNA may assist or interfere with the biological activity of this physiologically relevant DNA sequence.
Experimental
Materials
Coralyne (2), porphyrin 4 and thiazole orange (6) were commercially obtained and used without further purification (2: Acros Organics, Geel, Belgium; 4, 6: Fluka Chemie AG, Buchs, Switzerland). The [2.2.2]heptamethine cyanine dyes 1a–e [22], bis-quinolinium 3 [39] and tetraazoniahetarene 5 [23] were synthesized according to published procedures. Oligodeoxyribonucleotides (HPLC purified) a2 [d(ACAG4TGTG4ACAG4TGTG4)] and Fa2T [d(Fluo-ACAG4TGTG4ACAG4TGTG4-Tamra)] were purchased from Metabion Int. AG (Planegg/Martinsried). The ILPR-DNA a2 or Fa2T were dissolved in the proper buffer, heated to 95 °C for 5 min, cooled slowly over 4 hours to room temperature and used two days after storing. Potassium phosphate buffer: 25 mM K2HPO4, 70 mM KCl; adjusted with 25 mM KH2PO4 to pH 7.0; sodium cacodylate buffer: 10 mM Na(CH3)2AsO2·3H2O, 10 mM KCl, 90 mM LiCl; pH 7.2–7.3.
Equipment
Absorption spectroscopy: Varian Cary 100 Bio spectrophotometer; emission spectroscopy: Varian Cary Eclipse; CD spectroscopy: Chirascan CD-spectrometer, Applied Photophysics.
Methods
All photometric and fluorimetric titrations were performed in thermostated quartz cuvettes at 20 °C. Titrant solutions were freshly prepared by dilution from the stock solution. Spectrophotometer slit widths were 2 nm for photometric experiments and 5 nm for fluorimetric experiments. In fluorimetric experiments, the spectra were smoothed with implemented moving-average function by a factor of 5. The binding constants were determined by fitting the binding isotherms from the fluorimetric titrations to the established theoretical model according to the independent-site model (Equation 1) [31].
![[1860-5397-10-314-i2]](/bjoc/content/inline/1860-5397-10-314-i2.svg?max-width=590&scale=1.18182)
Where y is the normalized intensity; A = 1/Kb; B = cLig, and n is the number of the binding sites per quadruplex DNA.
Thermal denaturation experiments were performed according to the published procedure [20].
For Job plot analysis (continuous variations method), a series of samples was prepared with a constant sum of concentrations at 10.0 μM, but with varying concentrations of ligand and ILPR quadruplex. The fluorescence spectra were recorded for each sample with λex = 580 nm for cyanine derivative 1e, and λex = 431 nm for porphyrin derivative 4 and λex = 490 nm for thiazole orange (6). The maximum fluorescence intensity was plotted versus the molar fraction of the corresponding ligand, XLigand. For the determination of the maximum the ascending and descending segments of the curve were fitted to linear lines, respectively, and the intercept of both lines denotes the maximum and thus the stoichiometry of the complex.
For the CD experiments five samples were prepared with fixed ILPR-DNA concentration (cDNA = 20.0 μM). In four of the samples different amounts of ligand were added to obtain different ligand concentrations (5, 10, 20, 40 μM). CD signals were recorded with a band width of 1 nm, a recording speed of 1 nm s−1 and a time per data point of 0.5 s. For the temperature dependent circular dichroism (CD) experiments, solutions of the ILPR-quadruplex a2 (20 μM) and the ligand (20 μM) were prepared in potassium phosphate buffer (95 mM, pH 7.0) and measured after an equilibration time of 24 h. After each measurement, the temperature was increased by 5 °C or 10 °C, after the temperature has been kept to an equilibration time of 10 min.
Supporting Information
Figures of fluorimetric DNA denaturation experiments, photometric titration of a2 ILPR-DNA into ligands 1–6, photometric and fluorimetric titration of 22AG into ligands 1b–e, plots of the CD intensity change of a2 ILPR-DNA in presence of ligands 1–6 and CD spectra of 22AG in presence of ligands 1d, 1e and 6 are provided.
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Bell, G. I.; Horita, S.; Karam, J. H. Diabetes 1984, 33, 176–183. doi:10.2337/diab.33.2.176
Return to citation in text: [1] -
Kennedy, G. C.; German, M. S.; Rutter, W. J. Nat. Genet. 1995, 9, 293–298. doi:10.1038/ng0395-293
Return to citation in text: [1] [2] -
Rotwein, P.; Yokoyama, S.; Didier, D. K.; Chirgwin, J. M. Am. J. Hum. Genet. 1986, 39, 291–299.
Return to citation in text: [1] -
Catasti, P.; Chen, X.; Moyzis, R. K.; Bradbury, E. M.; Gupta, G. J. Mol. Biol. 1996, 264, 534–545. doi:10.1006/jmbi.1996.0659
Return to citation in text: [1] -
Lew, A.; Rutter, W. J.; Kennedy, G. C. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 12508–12512. doi:10.1073/pnas.97.23.12508
Return to citation in text: [1] -
Schonhoft, J. D.; Das, A.; Achamyeleh, F.; Samdani, S.; Sewell, A.; Mao, H.; Basu, S. Biopolymers 2010, 93, 21–31. doi:10.1002/bip.21289
Return to citation in text: [1] -
Gerasimov, J. Y.; Schaefer, C. S.; Yang, W.; Grout, R. L.; Lai, R. Y. Biosens. Bioelectron. 2013, 42, 62–68. doi:10.1016/j.bios.2012.10.046
Return to citation in text: [1] -
Xiao, J.; Carter, J. A.; Frederick, K. A.; McGown, L. B. J. Sep. Sci. 2009, 32, 1654–1664. doi:10.1002/jssc.200900060
Return to citation in text: [1] -
Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r
Return to citation in text: [1] [2] [3] [4] [5] -
De Messieres, M.; Chang, J.-C.; Brawn-Cinani, B.; La Porta, A. Phys. Rev. Lett. 2012, 109, 058101. doi:10.1103/PhysRevLett.109.058101
Return to citation in text: [1] -
Hammond-Kosack, M. C. U.; Dobrinski, B.; Lurz, R.; Docherty, K.; Kilpatrick, M. W. Nucleic Acids Res. 1992, 20, 231–236. doi:10.1093/nar/20.2.231
Return to citation in text: [1] -
Perkins, T. T.; Li, H.-W.; Dalal, R. V.; Gelles, J.; Block, S. M. Biophys. J. 2004, 86, 1640–1648. doi:10.1016/S0006-3495(04)74232-0
Return to citation in text: [1] -
Liu, J.-q.; Chen, C.-y.; Xue, Y.; Hao, Y.-h.; Tan, Z. J. Am. Chem. Soc. 2010, 132, 10521–10527. doi:10.1021/ja1038165
Return to citation in text: [1] -
Schonhoft, J. D.; Bajracharya, R.; Dhakal, S.; Yu, Z.; Mao, H.; Basu, S. Nucleic Acids Res. 2009, 37, 3310–3320. doi:10.1093/nar/gkp181
Return to citation in text: [1] -
Oganesian, L.; Bryan, T. M. BioEssays 2007, 29, 155–165. doi:10.1002/bies.20523
Return to citation in text: [1] -
Murat, P.; Singh, Y.; Defrancq, E. Chem. Soc. Rev. 2011, 40, 5293–5307. doi:10.1039/c1cs15117g
Return to citation in text: [1] -
Paramasivan, S.; Bolton, P. H. Nucleic Acids Res. 2008, 36, e106. doi:10.1093/nar/gkn487
Return to citation in text: [1] -
Yan, J.-W.; Chen, S.-B.; Liu, H.-Y.; Ye, W.-J.; Ou, T.-M.; Tan, J.-H.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Chem. Commun. 2014, 50, 6927–6930. doi:10.1039/c4cc01472c
Return to citation in text: [1] -
Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a
Return to citation in text: [1] [2] [3] [4] [5] -
Nanjunda, R.; Owens, E. A.; Mickelson, L.; Alyabyev, S.; Kilpatrick, N.; Wang, S.; Henary, M.; Wilson, W. D. Bioorg. Med. Chem. 2012, 20, 7002–7011. doi:10.1016/j.bmc.2012.10.008
Return to citation in text: [1] -
Reichardt, C.; Mormann, W. Chem. Ber. 1972, 105, 1815–1837. doi:10.1002/cber.19721050606
Return to citation in text: [1] [2] -
Jäger, K.; Bats, J. W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B. O. Chem. – Eur. J. 2012, 18, 10903–10915. doi:10.1002/chem.201103019
Return to citation in text: [1] [2] [3] [4] -
Pennarun, G.; Granotier, C.; Gauthier, L. R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.-F.; Mergny, J.-L.; Mailliet, P.; Boussin, F. D. Oncogene 2005, 24, 2917–2928. doi:10.1038/sj.onc.1208468
Return to citation in text: [1] -
Granzhan, A.; Ihmels, H.; Jäger, K. Chem. Commun. 2009, 1249–1251. doi:10.1039/b812891j
Return to citation in text: [1] -
Wang, P.; Leung, C.-H.; Ma, D.-L.; Yan, S.-C.; Che, C.-M. Chem. – Eur. J. 2010, 16, 6900–6911. doi:10.1002/chem.201000167
Return to citation in text: [1] -
Monchaud, D.; Allain, C.; Teulade-Fichou, M.-P. Bioorg. Med. Chem. Lett. 2006, 16, 4842–4845. doi:10.1016/j.bmcl.2006.06.067
Return to citation in text: [1] -
Williamson, J. R. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 703–730. doi:10.1146/annurev.bb.23.060194.003415
Return to citation in text: [1] -
Renčiuk, D.; Zhou, J.; Beaurepaire, L.; Guédin, A.; Bourdoncle, A.; Mergny, J.-L. Methods 2012, 57, 122–128. doi:10.1016/j.ymeth.2012.03.020
Return to citation in text: [1] -
Monchaud, D.; Allain, C.; Teulade-Fichou, M.-P. Nucleosides, Nucleotides Nucleic Acids 2007, 26, 1585–1588. doi:10.1080/15257770701548212
Return to citation in text: [1] -
Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j
Return to citation in text: [1] [2] -
Vorlíčková, M.; Kejnovská, I.; Sagi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Methods 2012, 57, 64–75. doi:10.1016/j.ymeth.2012.03.011
Return to citation in text: [1] [2] -
Allain, C.; Monchaud, D.; Teulade-Fichou, M.-P. J. Am. Chem. Soc. 2006, 128, 11890–11893. doi:10.1021/ja062193h
Return to citation in text: [1] -
Bhadra, K.; Kumar, G. S. Biochim. Biophys. Acta 2011, 1810, 485–496. doi:10.1016/j.bbagen.2011.01.011
Return to citation in text: [1] -
Paramasivan, S.; Rujan, I.; Bolton, P. H. Methods 2007, 43, 324–331. doi:10.1016/j.ymeth.2007.02.009
Return to citation in text: [1] -
Martino, L.; Pagano, B.; Fotticchia, I.; Neidle, S.; Giancola, C. J. Phys. Chem. B 2009, 113, 14779–14786. doi:10.1021/jp9066394
Return to citation in text: [1] -
Lim, K. W.; Amrane, S.; Bouaziz, S.; Xu, W.; Mu, Y.; Patel, D. J.; Luu, K. N.; Phan, A. T. J. Am. Chem. Soc. 2009, 131, 4301–4309. doi:10.1021/ja807503g
Return to citation in text: [1] -
Zhang, H.-J.; Wang, X.-F.; Wang, P.; Ai, X.-C.; Zhang, J.-P. Photochem. Photobiol. Sci. 2008, 7, 948–955. doi:10.1039/b809322a
See also for stabilization of anti-parallel quadruplex by 4.
Return to citation in text: [1] -
Dorazco-González, A.; Höpfl, H.; Medrano, F.; Yatsimirsky, A. K. J. Org. Chem. 2010, 75, 2259–2273. doi:10.1021/jo100037m
Return to citation in text: [1]
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 10. | Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r |
| 20. | Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a |
| 1. | Bell, G. I.; Horita, S.; Karam, J. H. Diabetes 1984, 33, 176–183. doi:10.2337/diab.33.2.176 |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 23. | Jäger, K.; Bats, J. W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B. O. Chem. – Eur. J. 2012, 18, 10903–10915. doi:10.1002/chem.201103019 |
| 24. | Pennarun, G.; Granotier, C.; Gauthier, L. R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.-F.; Mergny, J.-L.; Mailliet, P.; Boussin, F. D. Oncogene 2005, 24, 2917–2928. doi:10.1038/sj.onc.1208468 |
| 25. | Granzhan, A.; Ihmels, H.; Jäger, K. Chem. Commun. 2009, 1249–1251. doi:10.1039/b812891j |
| 26. | Wang, P.; Leung, C.-H.; Ma, D.-L.; Yan, S.-C.; Che, C.-M. Chem. – Eur. J. 2010, 16, 6900–6911. doi:10.1002/chem.201000167 |
| 27. | Monchaud, D.; Allain, C.; Teulade-Fichou, M.-P. Bioorg. Med. Chem. Lett. 2006, 16, 4842–4845. doi:10.1016/j.bmcl.2006.06.067 |
| 32. | Vorlíčková, M.; Kejnovská, I.; Sagi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Methods 2012, 57, 64–75. doi:10.1016/j.ymeth.2012.03.011 |
| 4. | Catasti, P.; Chen, X.; Moyzis, R. K.; Bradbury, E. M.; Gupta, G. J. Mol. Biol. 1996, 264, 534–545. doi:10.1006/jmbi.1996.0659 |
| 5. | Lew, A.; Rutter, W. J.; Kennedy, G. C. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 12508–12512. doi:10.1073/pnas.97.23.12508 |
| 6. | Schonhoft, J. D.; Das, A.; Achamyeleh, F.; Samdani, S.; Sewell, A.; Mao, H.; Basu, S. Biopolymers 2010, 93, 21–31. doi:10.1002/bip.21289 |
| 7. | Gerasimov, J. Y.; Schaefer, C. S.; Yang, W.; Grout, R. L.; Lai, R. Y. Biosens. Bioelectron. 2013, 42, 62–68. doi:10.1016/j.bios.2012.10.046 |
| 8. | Xiao, J.; Carter, J. A.; Frederick, K. A.; McGown, L. B. J. Sep. Sci. 2009, 32, 1654–1664. doi:10.1002/jssc.200900060 |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 28. | Williamson, J. R. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 703–730. doi:10.1146/annurev.bb.23.060194.003415 |
| 36. | Martino, L.; Pagano, B.; Fotticchia, I.; Neidle, S.; Giancola, C. J. Phys. Chem. B 2009, 113, 14779–14786. doi:10.1021/jp9066394 |
| 2. | Kennedy, G. C.; German, M. S.; Rutter, W. J. Nat. Genet. 1995, 9, 293–298. doi:10.1038/ng0395-293 |
| 3. | Rotwein, P.; Yokoyama, S.; Didier, D. K.; Chirgwin, J. M. Am. J. Hum. Genet. 1986, 39, 291–299. |
| 20. | Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a |
| 21. | Nanjunda, R.; Owens, E. A.; Mickelson, L.; Alyabyev, S.; Kilpatrick, N.; Wang, S.; Henary, M.; Wilson, W. D. Bioorg. Med. Chem. 2012, 20, 7002–7011. doi:10.1016/j.bmc.2012.10.008 |
| 34. | Bhadra, K.; Kumar, G. S. Biochim. Biophys. Acta 2011, 1810, 485–496. doi:10.1016/j.bbagen.2011.01.011 |
| 2. | Kennedy, G. C.; German, M. S.; Rutter, W. J. Nat. Genet. 1995, 9, 293–298. doi:10.1038/ng0395-293 |
| 22. | Reichardt, C.; Mormann, W. Chem. Ber. 1972, 105, 1815–1837. doi:10.1002/cber.19721050606 |
| 35. | Paramasivan, S.; Rujan, I.; Bolton, P. H. Methods 2007, 43, 324–331. doi:10.1016/j.ymeth.2007.02.009 |
| 14. | Liu, J.-q.; Chen, C.-y.; Xue, Y.; Hao, Y.-h.; Tan, Z. J. Am. Chem. Soc. 2010, 132, 10521–10527. doi:10.1021/ja1038165 |
| 16. | Oganesian, L.; Bryan, T. M. BioEssays 2007, 29, 155–165. doi:10.1002/bies.20523 |
| 17. | Murat, P.; Singh, Y.; Defrancq, E. Chem. Soc. Rev. 2011, 40, 5293–5307. doi:10.1039/c1cs15117g |
| 20. | Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 13. | Perkins, T. T.; Li, H.-W.; Dalal, R. V.; Gelles, J.; Block, S. M. Biophys. J. 2004, 86, 1640–1648. doi:10.1016/S0006-3495(04)74232-0 |
| 18. | Paramasivan, S.; Bolton, P. H. Nucleic Acids Res. 2008, 36, e106. doi:10.1093/nar/gkn487 |
| 19. | Yan, J.-W.; Chen, S.-B.; Liu, H.-Y.; Ye, W.-J.; Ou, T.-M.; Tan, J.-H.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Chem. Commun. 2014, 50, 6927–6930. doi:10.1039/c4cc01472c |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 10. | Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 11. | De Messieres, M.; Chang, J.-C.; Brawn-Cinani, B.; La Porta, A. Phys. Rev. Lett. 2012, 109, 058101. doi:10.1103/PhysRevLett.109.058101 |
| 12. | Hammond-Kosack, M. C. U.; Dobrinski, B.; Lurz, R.; Docherty, K.; Kilpatrick, M. W. Nucleic Acids Res. 1992, 20, 231–236. doi:10.1093/nar/20.2.231 |
| 23. | Jäger, K.; Bats, J. W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B. O. Chem. – Eur. J. 2012, 18, 10903–10915. doi:10.1002/chem.201103019 |
| 10. | Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r |
| 15. | Schonhoft, J. D.; Bajracharya, R.; Dhakal, S.; Yu, Z.; Mao, H.; Basu, S. Nucleic Acids Res. 2009, 37, 3310–3320. doi:10.1093/nar/gkp181 |
| 33. | Allain, C.; Monchaud, D.; Teulade-Fichou, M.-P. J. Am. Chem. Soc. 2006, 128, 11890–11893. doi:10.1021/ja062193h |
| 20. | Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 37. | Lim, K. W.; Amrane, S.; Bouaziz, S.; Xu, W.; Mu, Y.; Patel, D. J.; Luu, K. N.; Phan, A. T. J. Am. Chem. Soc. 2009, 131, 4301–4309. doi:10.1021/ja807503g |
| 38. |
Zhang, H.-J.; Wang, X.-F.; Wang, P.; Ai, X.-C.; Zhang, J.-P. Photochem. Photobiol. Sci. 2008, 7, 948–955. doi:10.1039/b809322a
See also for stabilization of anti-parallel quadruplex by 4. |
| 29. | Renčiuk, D.; Zhou, J.; Beaurepaire, L.; Guédin, A.; Bourdoncle, A.; Mergny, J.-L. Methods 2012, 57, 122–128. doi:10.1016/j.ymeth.2012.03.020 |
| 22. | Reichardt, C.; Mormann, W. Chem. Ber. 1972, 105, 1815–1837. doi:10.1002/cber.19721050606 |
| 39. | Dorazco-González, A.; Höpfl, H.; Medrano, F.; Yatsimirsky, A. K. J. Org. Chem. 2010, 75, 2259–2273. doi:10.1021/jo100037m |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 10. | Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 10. | Timmer, C. M.; Michmerhuizen, N. L.; Witte, A. B.; Van Winkle, M.; Zhou, D.; Sinniah, K. J. Phys. Chem. B 2014, 118, 1784–1790. doi:10.1021/jp411293r |
| 9. | Yu, Z.; Schonhoft, J. D.; Dhakal, S.; Bajracharya, R.; Hegde, R.; Basu, S.; Mao, H. J. Am. Chem. Soc. 2009, 131, 1876–1882. doi:10.1021/ja806782s |
| 31. | Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j |
| 20. | Ihmels, H.; Thomas, L. Org. Biomol. Chem. 2013, 11, 480–487. doi:10.1039/c2ob26779a |
| 32. | Vorlíčková, M.; Kejnovská, I.; Sagi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Methods 2012, 57, 64–75. doi:10.1016/j.ymeth.2012.03.011 |
| 23. | Jäger, K.; Bats, J. W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B. O. Chem. – Eur. J. 2012, 18, 10903–10915. doi:10.1002/chem.201103019 |
| 23. | Jäger, K.; Bats, J. W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B. O. Chem. – Eur. J. 2012, 18, 10903–10915. doi:10.1002/chem.201103019 |
| 30. | Monchaud, D.; Allain, C.; Teulade-Fichou, M.-P. Nucleosides, Nucleotides Nucleic Acids 2007, 26, 1585–1588. doi:10.1080/15257770701548212 |
| 31. | Stootman, F. H.; Fisher, D. M.; Rodger, A.; Aldrich-Wright, J. R. Analyst 2006, 131, 1145–1151. doi:10.1039/b604686j |
© 2014 Dzubiel et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)