Abstract
A mild and efficient methodology for the bromination of phenols and alkenes has been developed utilizing visible light-induced photoredox catalysis. The bromine was generated in situ from the oxidation of Br− by Ru(bpy)33+, both of which resulted from the oxidative quenching process.

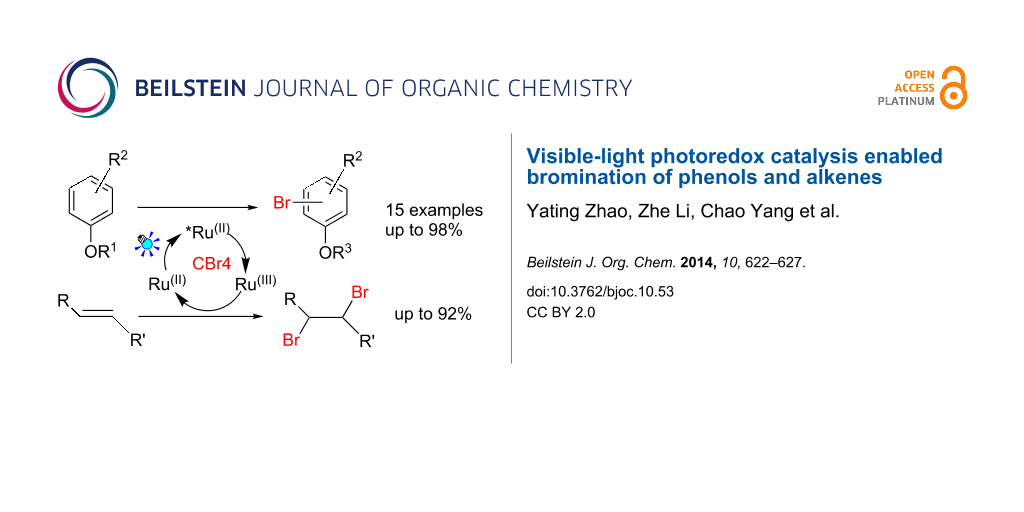
Graphical Abstract
Introduction
Bromophenols serve as important synthetic intermediates for a variety of naturally occurring biologically active compounds and are also important constituents of industrial chemicals [1-5]. Thus, numerous methods were developed for the electrophilic bromination of phenols. The typical approaches include direct electrophilic halogenation by using molecular bromine or N-bromosuccinimide (NBS) [6-8], organometallic catalyst-promoted bromination [9-12], and the oxidative bromination of phenols [13-15]. Nevertheless, most of the methods suffer from several drawbacks such as toxic reagents, harsh conditions, low yields, and low chemo- and regioselectivity. Hence, the development of an environmentally friendly methodology for the bromination of phenols with high chemoselectivity under mild and operationally simple conditions is still appealing.
Recently, an intriguing and promising strategy for the application of photoredox catalysts to initiate single electron transfer processes have been developed [16-22]. Since the pioneering work from the groups of MacMillan [23-25], Stephenson [26-28], Yoon [29-31] and others [32-44] demonstrated the usefulness of Ru(bpy)3Cl2 and its application to various visible-light-induced synthetic transformations, visible-light-photoredox catalysis has emerged as a growing field in organic chemistry and has been successfully applied in a variety of reactions. In the oxidative quenching process [45-47], Ru(bpy)32+ excited by visible light generates Ru(bpy)32+*, which was oxidized to Ru(bpy)33+ in the presence of oxidative quenchers. CBr4 is an example of a suitable oxidative quencher and leads to the formation of Br− and •CBr3. We envision that Ru(bpy)33+ is a strong oxidant (1.29 V vs SCE, in CH3CN) that could sequentially oxidize the resulting Br− (1.087 V vs SCE, in CH3CN) to generate the bromine for the bromination of phenols and other substrates in situ (Scheme 1), thus avoiding the use of highly toxic and volatile liquid bromine.
![[1860-5397-10-53-i1]](/bjoc/content/inline/1860-5397-10-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Strategy for in situ generation of bromine.
Scheme 1: Strategy for in situ generation of bromine.
Results and Discussion
Our initial investigation was carried out on protected 4-methoxyphenol 1a and CBr4 in dried CH3CN in the presence of Ru(bpy)3Cl2 (5.0 mol %) with visible light irradiation (blue LEDs, λmax = 435 nm) for 6 hours. The corresponding 2-bromo-4-methoxyphenol (2a) was obtained in 78% yield (Table 1, entry 1), whereas 3-bromo-4-methoxyphenol was not observed. The optimization of the reaction conditions were conducted by screening selected solvents and the amount of the photoredox catalyst using 1a as the representative substrate. As can be seen in Table 1, the solvent had a significant effect on the reaction efficiency. The reaction did not work well in DMF, MeOH, THF, CH2Cl2, EtOAc, CH3CN with 10 equivalents of H2O or 1,4-dioxane (Table 1, entries 5–11). The reaction in CH3CN and open to air led to the highest yield, 94% (Table 1, entry 4), whereas the reaction conducted under N2 or O2, or in DMSO and open to air gave lower yields (Table 1, entries 2, 3, 12). Our final optimization showed that the reaction also provided comparable results when the catalyst loading was reduced to 3% or 1% (Table 1, entry 13). It should be pointed out that an exclusion of either the photocatalyst or the light source did not afford the desired product 2a. Therefore, the reaction conditions of CBr4 (1 equiv) in dried CH3CN in the presence of Ru(bpy)3Cl2 (5.0 mol %) with visible light irradiation (blue LEDs, λmax = 435 nm) and open to air were utilized to test the scope of the reaction.
Table 1: Survey of the photocatalytic bromination reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-53-i5.svg?max-width=637&scale=1.0)
|
||
| entry | conditionsa | yield (%)b |
|---|---|---|
| 1 | CH3CN, tube closed | 78 |
| 2 | CH3CN, N2 | 89 |
| 3 | CH3CN, O2 | 46 |
| 4 | CH3CN, open to air | 94 |
| 5 | CH3OH, open to air | 23 |
| 6 | CH2Cl2, open to air | 0 |
| 7 | DMF, open to air | 63 |
| 8 | THF, open to air | 0 |
| 9 | EtOAc, open to air | 0 |
| 10 | 1,4-dioxane, open to air | 0 |
| 11 | CH3CN + 10 equiv H2O, open to air | 0 |
| 12 | DMSO, open to air | 82 |
| 13 | CH3CN, Ru(bpy)3Cl2 (3%), open to air | 86 |
aReaction conditions: substrate 1a (0.1 mmol), CBr4 (0.1 mmol), Ru(bpy)3Cl2 (5 mol %), solvent (0.1 M), blue LEDs (1W). bYields were determined by GC analysis.
With the optimized conditions in hand, we prepared a variety of phenols which were subjected to the photocatalytic reaction. In general, both electron-withdrawing and electron–donating groups were tolerated as substituents R2 in this process. Interestingly, the substrates protected with TMS (trimethylsilyl), TBS (tert-butyldimethylsilyl), MOM (methoxymethyl) and THP (tetrahydropyranyl) groups led to the corresponding bromophenols via a Tandem bromination/deprotection reaction (Table 2, entries 1–8, 12, 13, and 15), among which the cases with substituents at para- and ortho-position afforded 2- and 4-bromophenol, respectively, in good to excellent yields (Table 2, entries 1–5 and 12). The compound substituted with a methoxy group at the meta-position (1b) led to both 2- and 4-bromophenols 2b and 2b' with a ratio of 2:1 (Table 2, entry 8). Without any substituent at the phenol moiety mono- and dibromophenols were obtained with a ratio of 3:2 (Table 2, entries 6 and 7). Notably, 1-bromonaphthalen-2-ol and 1-bromo-2-methoxynaphthalene could be prepared in good yields with high regioselectivity from TMS and methyl protected naphthalen-2-ol (Table 2, entries 13 and 14). The direct treatment of 3-methoxyphenol under the same reaction conditions afforded 2- and 4-bromo-3-methoxyphenol with a ratio of 3:2 in a synthetically acceptable yield (Table 2, entry 11). Phenols protected with Bn or Ms groups led to 2- and 4-bromophenol derivatives in excellent yields without the loss of Bn or Ms groups (Table 2, entries 9 and 10).
Table 2: Scope of the photocatalytic bromination of phenols.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-53-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%)a |
|---|---|---|---|
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-53-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-53-i8.svg?max-width=637&scale=1.0)
|
Conditionsb | |
| 1 | R1 = TMS; R2 = OMe; R3 = H | 88 | |
| 2 | R1 = TMS; R2 = Me; R3 = H | 69 | |
| 3 | R1 = TMS; R2 = Cl; R3 = H | 58 | |
| 4 | R1 = TBS; R2 = OMe; R3 = H | 85 | |
| 5 | R1 = MOM; R2 = Me; R3 = H | 97 | |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-53-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-53-i10.svg?max-width=637&scale=1.0)
|
||
| 6 | R1 = TMS; R3 = H (3:2)c | 79 | |
| 7 | R1 = THP; R3 = H (3:2)c | 79 | |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-53-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-53-i12.svg?max-width=637&scale=1.0)
|
||
| 8 | R1 = TMS; R2 = OMe; R3 = H (2:1)c | 73 | |
| 9 | R1 = Ms; R2 = OMe; R3 = Ms (5:1)c | 95 | |
| 10 | R1 = Bn; R2 = OMe; R3 = Bn (5:3)c | 98 | |
| 11 | R1 = H; R2 = OMe; R3 = H (3:2)c | 40 | |
| 12 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-53-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-53-i14.svg?max-width=637&scale=1.0)
|
84 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-53-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-53-i16.svg?max-width=637&scale=1.0)
|
||
| 13 | R1 = TMS; R3 = H | 76 | |
| 14 | R1 = Me; R3 = Me | 98 | |
| 15 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-53-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-53-i18.svg?max-width=637&scale=1.0)
|
46 |
aIsolated yield based on complete consume of the starting material. bReaction conditions: substrate 1 (0.1 mmol), CBr4 (0.1 mmol), Ru(bpy)3Cl2 (5 mol %), dried CH3CN, blue LEDs (1 W), open to air. cRatio of the isomers in parentheses.
The bromination of phenols could be controlled by the amount of CBr4. For example, when TMS protected 3-methoxyphenol was treated with 2 equivalents of CBr4 under similar conditions (Table 2), a dibromophenol product was directly obtained in a high yield (95%) (Scheme 2), which also could be prepared from the same starting materials in two steps (Table 2).
![[1860-5397-10-53-i2]](/bjoc/content/inline/1860-5397-10-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of dibromophenol product 2b''.
Scheme 2: Synthesis of dibromophenol product 2b''.
We also conducted a control experiment by reacting stilbene with CBr4 (1 equiv) in dry CH3CN in the presence of Ru(bpy)3Cl2 (5.0 mol %) with visible-light irradiation (blue LEDs, λmax = 435 nm) for 72 hours, which led to the anti-1,2-dibromo-1,2-diphenylethane in 92% yield. This result is in accordance with the direct bromination of stilbene from liquid bromine [47]. Based on this result, our protocol provides an easily manageable and environment-friendly pathway to the bromination of alkenes. We further examined the scope of the reaction, and the results are summarized in Scheme 3. The 1,2-dibromo products were obtained in moderate to high yields.
![[1860-5397-10-53-i3]](/bjoc/content/inline/1860-5397-10-53-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the photocatalytic bromination of alkenes.
Scheme 3: Scope of the photocatalytic bromination of alkenes.
With the success of the bromination of phenols and alkenes, we further focused on the complementary bromination of diketones and cyclization reactions. The treatment of cyclohexane-1,3-dione (5) and (E)-4-(4-methoxyphenyl)but-3-en-1-ol (7) under the same reaction conditions led to 2,2-dibromocyclohexane-1,3-dione (6) and bromofuran compound 8 in 22% and 52% yield, respectively (Scheme 4). This outcome demonstrates the efficiency of the Ru(bpy)3Cl2/CBr4 photocatalytic system. The stereochemistry of the bromofuran compound was determined by 2D NMR spectra.
![[1860-5397-10-53-i4]](/bjoc/content/inline/1860-5397-10-53-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Bromination of diketones and cyclization reactions.
Scheme 4: Bromination of diketones and cyclization reactions.
Conclusion
In summary, we have developed a mild and operationally simple method for the in situ preparation of bromine utilizing a visible-light photoredox catalyst. The reaction proceeds with high chemical yield and regioselectivity for the bromination of phenols and alkenes. Further development of photoredox catalysis in the context of radical chemistry and its application in other reactions are currently underway in our laboratory.
Experimental
General procedure for the bromination of phenols and alkenes
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-53-i19.svg?max-width=637&scale=1.18182)
To a 10 mL round bottom flask equipped with a magnetic stir bar were added phenols or alkenes (0.1 mmol), CBr4 (33 mg, 0.1 mmol), dry CH3CN (1 mL) and Ru(bpy)3Cl2 (3.8 mg, 0.005 mmol). The mixture was irradiated with blue LEDs (1 W) at room temperature open to air until the starting material disappeared completely (monitored by TLC). After the reaction was completed, the solvent was concentrated in vacuo. The residue was purified by flash column chromatography to give the final product.
Bromination of diketones
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-53-i20.svg?max-width=637&scale=1.18182)
To a 10 mL round bottom flask equipped with a magnetic stir bar were added 5 (0.4 mmol), CBr4 (133 mg, 0.4 mmol), dry CH3CN (2 mL) and Ru(bpy)3Cl2 (15 mg, 0.02 mmol). The mixture was irradiated with blue LEDs (1 W) at room temperature open to air until the starting material was largely consumed (monitored by TLC). After the reaction was completed the solvent was concentrated in vacuo. The residue was purified by flash column chromatography to give the final product 6.
Synthesis of bromofuran
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-53-i21.svg?max-width=637&scale=1.18182)
To a 10 mL round bottom flask equipped with a magnetic stir bar were added 7 (0.13 mmol), CBr4 (43 mg, 0.13 mmol), LiBr (11 mg, 0.13 mmol), dry CH3CN (1 mL) and Ru(bpy)3Cl2 (4.5 mg, 0.006 mmol). The mixture was irradiated with blue LEDs (1 W) at room temperature open to air until the starting material disappeared completely (monitored by TLC). After the reaction was completed the solvent was concentrated in vacuo. The residue was purified by flash column chromatography to give the final product 8.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra for products. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937–1944. doi:10.1021/cr00021a014
Return to citation in text: [1] -
Heck, R. F. Org. React. 1982, 27, 345–390.
Return to citation in text: [1] -
Stille, J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508–524. doi:10.1002/anie.198605081
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Ma, D.; Cai, Q. Acc. Chem. Res. 2008, 41, 1450–1460. doi:10.1021/ar8000298
Return to citation in text: [1] -
Mitchell, R. H.; Lai, Y.-H.; Williams, R. V. J. Org. Chem. 1979, 44, 4733–4735. doi:10.1021/jo00393a066
Return to citation in text: [1] -
Carreno, M. C.; Garcia Ruano, J. L.; Sanz, G.; Toledo, M. A.; Urbano, A. J. Org. Chem. 1995, 60, 5328–5331. doi:10.1021/jo00121a064
Return to citation in text: [1] -
Park, M. Y.; Yang, S. G.; Jadhav, V.; Kim, Y. H. Tetrahedron Lett. 2004, 45, 4887–4890. doi:10.1016/j.tetlet.2004.04.112
Return to citation in text: [1] -
Wan, X.; Ma, Z.; Li, B.; Zhang, K.; Cao, S.; Zhang, S.; Shi, Z. J. Am. Chem. Soc. 2006, 128, 7416–7417. doi:10.1021/ja060232j
Return to citation in text: [1] -
Song, B.; Zheng, X.; Mo, J.; Xu, B. Adv. Synth. Catal. 2010, 352, 329–335. doi:10.1002/adsc.200900778
Return to citation in text: [1] -
Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215–5219. doi:10.1002/anie.200705613
Return to citation in text: [1] -
Castro, C. E.; Gaughan, E. J.; Owsley, D. C. J. Org. Chem. 1965, 30, 587–592. doi:10.1021/jo01013a069
Return to citation in text: [1] -
Bora, U.; Bose, G.; Chaudhuri, M. K.; Dhar, S. S.; Gopinath, R.; Khan, A. T.; Patel, B. K. Org. Lett. 2000, 2, 247–249. doi:10.1021/ol9902935
Return to citation in text: [1] -
Narender, N.; Mohan, K. V. V. K.; Reddy, R. V.; Srinivasu, P.; Kulkarni, S. J.; Raghavan, K. V. J. Mol. Catal. A: Chem. 2003, 192, 73–77. doi:10.1016/S1381-1169(02)00131-0
Return to citation in text: [1] -
Adibi, H.; Hajipour, A. R.; Hashemi, M. Tetrahedron Lett. 2007, 48, 1255–1259. doi:10.1016/j.tetlet.2006.12.033
Return to citation in text: [1] -
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687
Return to citation in text: [1] -
Inagaki, A.; Akita, M. Coord. Chem. Rev. 2010, 254, 1220–1239. doi:10.1016/j.ccr.2009.11.003
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] -
Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x
Return to citation in text: [1] -
McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920
Return to citation in text: [1] -
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976
Return to citation in text: [1] -
Nagib, D. A.; MacMillan, D. W. C. Nature 2011, 480, 224–228. doi:10.1038/nature10647
Return to citation in text: [1] -
Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y
Return to citation in text: [1] -
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582
Return to citation in text: [1] -
Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949
Return to citation in text: [1] -
Ischay, M. A.; Lu, Z.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 8572–8574. doi:10.1021/ja103934y
Return to citation in text: [1] -
Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f
Return to citation in text: [1] -
Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2009, 131, 14604–14605. doi:10.1021/ja903732v
Return to citation in text: [1] -
Tucker, J. W.; Narayanam, J. M. R.; Krabbe, S. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 368–371. doi:10.1021/ol902703k
Return to citation in text: [1] -
Furst, L.; Matsuura, B. S.; Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 3104–3107. doi:10.1021/ol101146f
Return to citation in text: [1] -
Maji, T.; Karmakar, A.; Reiser, O. J. Org. Chem. 2011, 76, 736–739. doi:10.1021/jo102239x
Return to citation in text: [1] -
Lu, Z.; Shen, M.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 1162–1164. doi:10.1021/ja107849y
Return to citation in text: [1] -
Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a
Return to citation in text: [1] -
Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m
Return to citation in text: [1] -
Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a
Return to citation in text: [1] -
Xie, J.; Xue, Q.; Jin, H.; Li, J.; Cheng, Y.; Zhu, C. Chem. Sci. 2013, 4, 1281–1286. doi:10.1039/c2sc22131d
Return to citation in text: [1] -
Liu, Q.; Li, Y.-N.; Zhang, H.-H.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Chem.–Eur. J. 2012, 18, 620–627. doi:10.1002/chem.201102299
Return to citation in text: [1] -
Chen, M.; Huang, Z.-T.; Zheng, Q.-Y. Chem. Commun. 2012, 48, 11686–11688. doi:10.1039/c2cc36866h
Return to citation in text: [1] -
Cai, S.; Zhao, X.; Wang, X.; Liu, Q.; Li, Z.; Wang, D. Z. Angew. Chem., Int. Ed. 2012, 51, 8050–8053. doi:10.1002/anie.201202880
Return to citation in text: [1] -
Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306
Return to citation in text: [1] -
Shih, H.-W.; Vander Wal, M. N.; Grange, R. L.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 13600–13603. doi:10.1021/ja106593m
Return to citation in text: [1] -
Nguyen, J. D.; Tucker, J. W.; Konieczynska, M. D.; Stephenson, C. R. J. J. Am. Chem. Soc. 2011, 133, 4160–4163. doi:10.1021/ja108560e
Return to citation in text: [1] -
Wallentin, C.-J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. J. Am. Chem. Soc. 2012, 134, 8875–8884. doi:10.1021/ja300798k
Return to citation in text: [1] -
Ma, K.; Li, S.; Weiss, R. G. Org. Lett. 2008, 10, 4155–4158. doi:10.1021/ol801327n
Return to citation in text: [1] [2]
| 1. | Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937–1944. doi:10.1021/cr00021a014 |
| 2. | Heck, R. F. Org. React. 1982, 27, 345–390. |
| 3. | Stille, J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508–524. doi:10.1002/anie.198605081 |
| 4. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 5. | Ma, D.; Cai, Q. Acc. Chem. Res. 2008, 41, 1450–1460. doi:10.1021/ar8000298 |
| 16. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 17. | Inagaki, A.; Akita, M. Coord. Chem. Rev. 2010, 254, 1220–1239. doi:10.1016/j.ccr.2009.11.003 |
| 18. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 19. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 20. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 21. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 22. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 13. | Bora, U.; Bose, G.; Chaudhuri, M. K.; Dhar, S. S.; Gopinath, R.; Khan, A. T.; Patel, B. K. Org. Lett. 2000, 2, 247–249. doi:10.1021/ol9902935 |
| 14. | Narender, N.; Mohan, K. V. V. K.; Reddy, R. V.; Srinivasu, P.; Kulkarni, S. J.; Raghavan, K. V. J. Mol. Catal. A: Chem. 2003, 192, 73–77. doi:10.1016/S1381-1169(02)00131-0 |
| 15. | Adibi, H.; Hajipour, A. R.; Hashemi, M. Tetrahedron Lett. 2007, 48, 1255–1259. doi:10.1016/j.tetlet.2006.12.033 |
| 9. | Wan, X.; Ma, Z.; Li, B.; Zhang, K.; Cao, S.; Zhang, S.; Shi, Z. J. Am. Chem. Soc. 2006, 128, 7416–7417. doi:10.1021/ja060232j |
| 10. | Song, B.; Zheng, X.; Mo, J.; Xu, B. Adv. Synth. Catal. 2010, 352, 329–335. doi:10.1002/adsc.200900778 |
| 11. | Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215–5219. doi:10.1002/anie.200705613 |
| 12. | Castro, C. E.; Gaughan, E. J.; Owsley, D. C. J. Org. Chem. 1965, 30, 587–592. doi:10.1021/jo01013a069 |
| 6. | Mitchell, R. H.; Lai, Y.-H.; Williams, R. V. J. Org. Chem. 1979, 44, 4733–4735. doi:10.1021/jo00393a066 |
| 7. | Carreno, M. C.; Garcia Ruano, J. L.; Sanz, G.; Toledo, M. A.; Urbano, A. J. Org. Chem. 1995, 60, 5328–5331. doi:10.1021/jo00121a064 |
| 8. | Park, M. Y.; Yang, S. G.; Jadhav, V.; Kim, Y. H. Tetrahedron Lett. 2004, 45, 4887–4890. doi:10.1016/j.tetlet.2004.04.112 |
| 32. | Tucker, J. W.; Narayanam, J. M. R.; Krabbe, S. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 368–371. doi:10.1021/ol902703k |
| 33. | Furst, L.; Matsuura, B. S.; Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 3104–3107. doi:10.1021/ol101146f |
| 34. | Maji, T.; Karmakar, A.; Reiser, O. J. Org. Chem. 2011, 76, 736–739. doi:10.1021/jo102239x |
| 35. | Lu, Z.; Shen, M.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 1162–1164. doi:10.1021/ja107849y |
| 36. | Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a |
| 37. | Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m |
| 38. | Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a |
| 39. | Xie, J.; Xue, Q.; Jin, H.; Li, J.; Cheng, Y.; Zhu, C. Chem. Sci. 2013, 4, 1281–1286. doi:10.1039/c2sc22131d |
| 40. | Liu, Q.; Li, Y.-N.; Zhang, H.-H.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Chem.–Eur. J. 2012, 18, 620–627. doi:10.1002/chem.201102299 |
| 41. | Chen, M.; Huang, Z.-T.; Zheng, Q.-Y. Chem. Commun. 2012, 48, 11686–11688. doi:10.1039/c2cc36866h |
| 42. | Cai, S.; Zhao, X.; Wang, X.; Liu, Q.; Li, Z.; Wang, D. Z. Angew. Chem., Int. Ed. 2012, 51, 8050–8053. doi:10.1002/anie.201202880 |
| 43. | Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306 |
| 44. | Shih, H.-W.; Vander Wal, M. N.; Grange, R. L.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 13600–13603. doi:10.1021/ja106593m |
| 47. | Ma, K.; Li, S.; Weiss, R. G. Org. Lett. 2008, 10, 4155–4158. doi:10.1021/ol801327n |
| 29. | Ischay, M. A.; Lu, Z.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 8572–8574. doi:10.1021/ja103934y |
| 30. | Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f |
| 31. | Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2009, 131, 14604–14605. doi:10.1021/ja903732v |
| 26. | Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y |
| 27. | Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582 |
| 28. | Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949 |
| 23. | McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920 |
| 24. | Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976 |
| 25. | Nagib, D. A.; MacMillan, D. W. C. Nature 2011, 480, 224–228. doi:10.1038/nature10647 |
| 45. | Nguyen, J. D.; Tucker, J. W.; Konieczynska, M. D.; Stephenson, C. R. J. J. Am. Chem. Soc. 2011, 133, 4160–4163. doi:10.1021/ja108560e |
| 46. | Wallentin, C.-J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. J. Am. Chem. Soc. 2012, 134, 8875–8884. doi:10.1021/ja300798k |
| 47. | Ma, K.; Li, S.; Weiss, R. G. Org. Lett. 2008, 10, 4155–4158. doi:10.1021/ol801327n |
© 2014 Zhao et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)