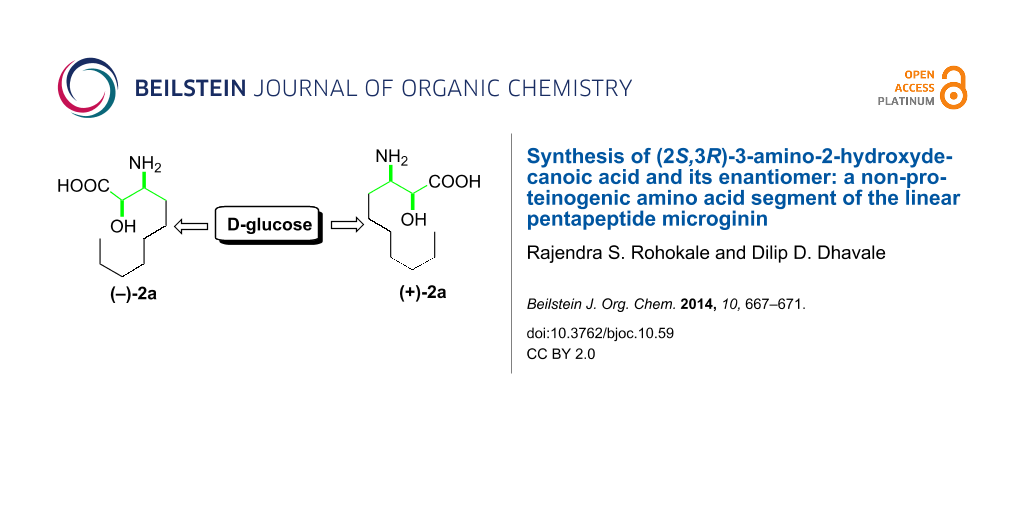
Abstract
A directed manipulation of the functional groups at C3 and C4 of D-glucose was demonstrated to synthesize naturally occurring (2S,3R)-α-hydroxy-β-aminodecanoic acid (AHDA, 2a) and its enantiomer 2b. The enantiomer of 2a is the N-terminal part of the natural linear pentapeptide microginin, which is used as an antihypertensive agent.

Graphical Abstract
Introduction
Microginin 1 (Figure 1), isolated from the cyanobacterium Microcystis aeruginosa, is a linear pentapeptide consisting of L-Tyr-L-N-Me-Tyr-L-Val-L-Ala and (2S,3R)-α-hydroxy-β-aminodecanoic acid ((2S,3R)-AHDA, 2a) [1]. Microginin is used as a hypertensive agent based on its biological activity against angiotensin converting enzyme, which is responsible for the vasoconstriction of blood vessels [2-4]. Amongst different amino acids present in microginin, (2S,3R)-AHDA (2a) is a non-proteinogenic natural amino acid attached at the N-terminal part of the peptide chain. The α-hydroxy-β-amino acid fragment in AHDA 2a is also present in linear peptides such as bestatin and valinoctin [5-8], which are isolated from the same species. In addition, the chiral α-hydroxy-β-amino acid constituent is an important component of protein kinase inhibitor compounds like balanol and the anticancer drug taxol [9].
![[1860-5397-10-59-1]](/bjoc/content/figures/1860-5397-10-59-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Microginin (1) and (2S,3R)-AHDA (2a).
Figure 1: Microginin (1) and (2S,3R)-AHDA (2a).
Due to the biological importance of (2S,3R)-AHDA, the enantioselective synthesis of its chiral core is a challenge task. This fact led to several approaches for the stereoselective synthesis of (2S,3R)-AHDA including (i) the enantioselective introduction of amino- and hydroxy-groups to olefinic acid by either asymmetric epoxidation, dihydroxylation or aminohydroxylation [10-15], (ii) the asymmetric synthesis of β-lactams by a Staudinger reaction between ketene and imine to give the corresponding amino acids [16], and (iii) the Lewis acid catalyzed multicomponent condensation reactions of aldehyde, an amine and ketene silyl acetal derivatives to get the vicinal hydroxylamino acids [17]. In addition, a few strategies employ a chiral pool approach. For example, Wee et al. utilized the zinc-silver-mediated reductive elimination of α-D-lyxofuranosyl phenylsulfone to get (4S,5S)-4-formyl-5-vinyl-2-oxazolidone, which was converted into 2a [18]. Merrer and co-workers used D-isoascorbic acid, which was transformed via (2R)-amino-1,3,4-triol to 2a [19]. Bergmeier et al. synthesized a chiral allyl alcohol from D-mannitol, which is converted to the azidoformate and thermally cyclized to a bicyclic aziridine. The opening of the aziridine with organocuprate led to a corresponding chiral hydroxylated amino acid core [20]. Although a number of chiron approaches are known [21], there is no report from D-glucose towards the synthesis of (2S,3R)-AHDA (2a) and its enantiomer (2R,3S)-AHDA (2b). As a part of our continuous interest in the synthesis of chiral amino acids [22,23] and their utility in the synthesis of iminosugars [24-29], we report here an efficient and practical approach for the synthesis of both enantiomers of AHDA (2a and 2b) from the same precursor D-glucose by simple manipulation of the functional groups.
We visualized that the structural and the stereochemical symmetry of both enantiomers (2a/2b) is present in D-glucose. The C1-carboxyl carbon atom of 2a is present at the C2 of the D-glucose, and the C4 carbon atom with an alkyl chain in 2a could be built on C5 of the D-glucose (Scheme 1). The required relative stereochemistry of the vicinal hydroxyamino functionality in 2a at C2 and C3 is embedded at the C3 and C4 of D-glucose, respectively, and needs to be manipulated by usual functional group transformations. Thus, for the synthesis of enantiomers 2a and 2b the corresponding sugar precursors were found to be suitably protected β-L-arabino-pentodialdo-1,4-furanose 3a [30,31] and α-D-ribo-pentodialdo-1,4-furanose 3b [32]. There exists a distinct possibility to synthesize these chiron synthons 3a and 3b from the easily available and cheap starting material D-glucose. Our results of the synthesis of both enantiomers 2a and 2b are described herein.
![[1860-5397-10-59-i1]](/bjoc/content/inline/1860-5397-10-59-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of AHDA.
Scheme 1: Retrosynthetic analysis of AHDA.
Results and Discussion
As reported earlier, D-glucose was converted to the 3-O-benzyl-1,2-O-isopropylidene-β-L-arabino-pentodialdo-1,4-furanose (3a) in 72% yield (Scheme 2) [30]. While targeting the synthesis of 2a, the Wittig olefination of 3a with n-hexyltriphenylphosphonium bromide and t-BuOK gave olefin 4a as a diasteromeric mixture of Z and E-isomers in the ratio 9.5:0.5 as shown by 1H NMR of the crude product. The catalytic hydrogenation of alkene 4a with 10% Pd/C in methanol:ethyl acetate (3:2) at balloon pressure gave 4-heptyl-L-threose derivative 5a as a viscous oil in 99% yield [33]. Removal of the 1,2-acetonide group with TFA–water in 5a provided an anomeric mixture of the hemiacetal, which was directly subjected to oxidative cleavage by using sodium metaperiodate in acetone–water (to cleave the anomeric carbon) followed by a treatment with sodium borohydride to give triol 6a as a viscous oil in 78% overall yield in three steps [34]. The primary hydroxy group of triol 6a was selectively monosilylated with t-butyldiphenylsilyl chloride to give 7a. Subsequently, the secondary hydroxy group in 7a was converted to azido derivative 8a with an inversion of the configuration by using diphenylphosphoryl azide in the presence of DBU in 88% yield [35]. Cleavage of the silyl functionality in 8a with n-tetrabutylammonium fluoride offered azido alcohol 9a as a viscous oil. The azido alcohol 9a was oxidized to the corresponding acid by using RuCl3·3H2O/NaIO4 to give 10a [36].
![[1860-5397-10-59-i2]](/bjoc/content/inline/1860-5397-10-59-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Finally, cleavage of the 2-O-benzyl ether and reduction of the 3-azido group to the corresponding amine in one step with 10% Pd/C in methanol provided (−)-α-hydroxy-β-aminodecanoic acid (AHDA, 2a) in 80% yield as a white solid. The spectral and analytical data of 2a was found to be in good agreement with published data ([α]D25 +5.6 (c 0.51, 1 M HCl). [α]D22 +7.3 (c 0.37, 1 M HCl)) [18].
The synthesis of AHDA enantiomer 2b was accomplished starting from 3-O-benzyl-1,2-O-isopropylidene-α-D-ribo-pentodialdo-1,4-furanose (3b) which was obtained from D-glucose in good yield as reported earlier [33]. Thus, the Wittig reaction of 3b followed by hydrogenation (10% Pd/C) gave 4-heptyl-D-threose derivative 5b (Scheme 3). Hydrolysis of 1,2-O-isopropylidene (TFA:H2O) followed by oxidative cleavage of the hemiacetal with NaIO4 and reduction with NaBH4 gave triol 6b,which was monosilylated with TBDPSCl to give 7b. Conversion of the secondary hydroxy group in 7b to azide 8b according to the Mitsunobu protocol, and deprotection followed by oxidation of the primary hydroxy group gave azido acid 10b. Finally, hydrogenolysis of the benzyl group and reduction of the azido group by using 10% Pd/C, in one pot, gave 2b in 30.1% overall yield from 3b. The spectral and analytical data was found to be in good agreement with reported data ([α]D30 −5.1(c 0.51, 1 M HCl). [α]D30 −6.2 (c 0.4, 1 M HCl)) [21].
![[1860-5397-10-59-i3]](/bjoc/content/inline/1860-5397-10-59-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we demonstrated a practical approach for the synthesis of both enantiomers of AHDA (2a and 2b) to obtain the stereochemistry required for the α-hydroxy-β-amino acid. Our method starts from D-glucose by an easy manipulation of its functional groups at C3 and C4. In addition, the chiral core (α-hydroxy-β-amino acid) in 2a is present in several biologically active compounds such as taxol, balanol and bestatin. Therefore, this methodology could be potentially exploited for the synthesis of the chiral segment of these compounds.
References
-
Okino, T.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Tetrahedron Lett. 1993, 34, 501–504. doi:10.1016/0040-4039(93)85112-A
Return to citation in text: [1] -
Wyvratt, M. J.; Patchett, A. A. Med. Res. Rev. 1985, 5, 483–531. doi:10.1002/med.2610050405
Return to citation in text: [1] -
Moore, R. E.; Banarjee, S.; Bomemann, V.; Caplan, F. R.; Chen, J. L.; Corley, D. G.; Larsen, L. K.; Moore, B. S.; Patterson, G. M. L.; Paul, V. J.; Stewart, J. B.; Williams, D. E. Pure Appl. Chem. 1989, 61, 521–524. doi:10.1351/pac198961030521
Return to citation in text: [1] -
Carmichael W. W. Handbook of Natural Toxins; Tu, A. T.; Ed.; Marcel Dekker: New York, 1988; pp. 121 ff.
Return to citation in text: [1] -
Umezawa, H.; Aoyagi, T.; Suda, H.; Hamada, M.; Takeuchi, T. J. Antibiot. 1976, 29, 97–99. doi:10.7164/antibiotics.29.97
Return to citation in text: [1] -
Nakamura, H.; Suda, H.; Takita, T.; Aoyagi, T.; Umezawa, H. J. Antibiot. 1976, 29, 102–103. doi:10.7164/antibiotics.29.102
Return to citation in text: [1] -
Sekizawa, R.; Iinuma, H.; Muraoka, Y.; Naganawa, H.; Kinoshita, N.; Nakamura, H.; Hamada, M.; Takeuchi, T.; Umezawa, K. J. Nat. Prod. 1996, 59, 232–236. doi:10.1021/np960067t
Return to citation in text: [1] -
Tsuda, M.; Muraoka, Y.; Takeuchi, T. J. Antibiot. 1996, 49, 1031–1035. doi:10.7164/antibiotics.49.1031
Return to citation in text: [1] -
Nicolau, K. C.; Dai, W.-M.; Guy, R. K. Angew. Chem., Int. Ed. 1994, 33, 15–44. doi:10.1002/anie.199400151
Return to citation in text: [1] -
Bunnage, M. E.; Burke, A. J.; Davies, S. G.; Goodwin, C. J. Tetrahedron: Asymmetry 1994, 5, 203–206. doi:10.1016/S0957-4166(00)86173-X
Return to citation in text: [1] -
Li, G.; Chang, H.-T.; Sharpless, K. B. Angew. Chem., Int. Ed. 1996, 35, 451–454. doi:10.1002/anie.199604511
Return to citation in text: [1] -
Chandrasekhar, S.; Mohapatra, S.; Yadav, J. S. Tetrahedron 1997, 8, 4089–4099. doi:10.1016/S0957-4166(97)00595-8
Return to citation in text: [1] -
Sugimura, H.; Miura, M.; Yamada, N. Tetrahedron: Asymmetry 1997, 8, 4089–4099. doi:10.1016/S0957-4166(97)00595-8
Return to citation in text: [1] -
Righi, G.; Chionne, A.; D’Achille, R.; Bonini, C. Tetrahedron: Asymmetry 1997, 8, 903–907. doi:10.1016/S0957-4166(97)00056-6
Return to citation in text: [1] -
Jeffords, C. W.; Mcnulty, J.; Lu, Z. H.; Wang, J. B. Helv. Chim. Acta 1996, 79, 1203–1216. doi:10.1002/hlca.19960790426
Return to citation in text: [1] -
Ha, H. J.; Ahn, Y. G.; Woo, J. S.; Lee, G. S.; Lee, W. K. Bull. Chem. Soc. Jpn. 2001, 74, 1667–1672. doi:10.1246/bcsj.74.1667
Return to citation in text: [1] -
Gassa, F.; Contini, A.; Fontana, G.; Pellegrino, S.; Gelmi, M. L. J. Org. Chem. 2010, 75, 7099–7106. doi:10.1021/jo1011762
Return to citation in text: [1] -
Wee, A. G. H.; McLeod, D. D. J. Org. Chem. 2003, 68, 6268–6273. doi:10.1021/jo034334t
Return to citation in text: [1] [2] -
Tuch, A.; Saniere, M.; Merrer, Y. L.; Depezay, J.-C. Tetrahedron: Asymmetry 1996, 7, 2901–2909. doi:10.1016/0957-4166(96)00381-3
Return to citation in text: [1] -
Bergmeier, S. C.; Stanchina, D. M. J. Org. Chem. 1999, 64, 2852–2859. doi:10.1021/jo9823893
Return to citation in text: [1] -
Shirode, N. M.; Deshmukh, A. R. A. S. Tetrahedron 2006, 62, 4615–4621. doi:10.1016/j.tet.2006.01.082
Return to citation in text: [1] [2] -
Kalamkar, N. B.; Kasture, V. M.; Dhavale, D. D. Tetrahedron Lett. 2010, 51, 6745–6747. doi:10.1016/j.tetlet.2010.10.086
Return to citation in text: [1] -
Kalamkar, N. B.; Kasture, V. M.; Dhavale, D. D. J. Org. Chem. 2008, 73, 3619–3622. doi:10.1021/jo702749r
Return to citation in text: [1] -
Dhavale, D. D.; Markad, S. D.; Karanjule, N. S.; Prakasha Reddy, J. J. Org. Chem. 2004, 69, 4760–4766. doi:10.1021/jo049509t
Return to citation in text: [1] -
Karanjule, N. S.; Markad, S. D.; Dhavale, D. D. J. Org. Chem. 2006, 71, 6273–6276. doi:10.1021/jo060823s
Return to citation in text: [1] -
Karanjule, N. S.; Markad, S. D.; Shinde, V. S.; Dhavale, D. D. J. Org. Chem. 2006, 71, 4667–4670. doi:10.1021/jo0601617
Return to citation in text: [1] -
Dhavale, D. D.; Ajish Kumar, K. S.; Chaudhari, V. D.; Sharma, T.; Sabharwal, S. G.; Prakasha, R. Org. Biomol. Chem. 2005, 3, 3720–3726. doi:10.1039/b509216g
Return to citation in text: [1] -
Ajish Kumar, K. S.; Chaudhari, V. D.; Puranik, V. G.; Dhavale, D. D. Eur. J. Org. Chem. 2007, 4895–4901. doi:10.1002/ejoc.200700461
Return to citation in text: [1] -
Pawar, N. J.; Parihar, V.; Chavan, S.; Joshi, R.; Joshi, P. V.; Sabharwal, S. G.; Puranik, V. G.; Dhavale, D. D. J. Org. Chem. 2012, 77, 7873–7882. doi:10.1021/jo3009534
Return to citation in text: [1] -
Sato, K.-i.; Akai, S.; Sakuma, M.; Kojima, M.; Suzuki, K.-j. Tetrahedron Lett. 2003, 44, 4903–4907. doi:10.1016/S0040-4039(03)01098-0
Return to citation in text: [1] [2] -
Hanessian, S. Total Synthesis of Natural Products: The "Chiron" Approach; Pergamon Press: Oxford, 1983.
Return to citation in text: [1] -
Patil, N. T.; John, S.; Sabharwal, S. G.; Dhavale, D. D. Bioorg. Med. Chem. 2002, 10, 2155–2160. doi:10.1016/S0968-0896(02)00073-1
Return to citation in text: [1] -
Bindra, J.; Grodski, A. J. Org. Chem. 1978, 43, 3240. doi:10.1021/jo00410a031
Return to citation in text: [1] [2] -
Mane, R. S.; Ajish Kumar, K. S.; Dhavale, D. D. J. Org. Chem. 2008, 73, 3284–3287. doi:10.1021/jo800044r
Return to citation in text: [1] -
Thompson, A. S.; Humphrey, G. R.; DeMarco, A. M.; Marthe, D. J.; Grabowaski, E. J. J. J. Org. Chem. 1993, 58, 5886–5888. doi:10.1021/jo00074a008
Return to citation in text: [1] -
Carlsen, P. J. H.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045
Return to citation in text: [1]
| 35. | Thompson, A. S.; Humphrey, G. R.; DeMarco, A. M.; Marthe, D. J.; Grabowaski, E. J. J. J. Org. Chem. 1993, 58, 5886–5888. doi:10.1021/jo00074a008 |
| 33. | Bindra, J.; Grodski, A. J. Org. Chem. 1978, 43, 3240. doi:10.1021/jo00410a031 |
| 34. | Mane, R. S.; Ajish Kumar, K. S.; Dhavale, D. D. J. Org. Chem. 2008, 73, 3284–3287. doi:10.1021/jo800044r |
| 1. | Okino, T.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Tetrahedron Lett. 1993, 34, 501–504. doi:10.1016/0040-4039(93)85112-A |
| 10. | Bunnage, M. E.; Burke, A. J.; Davies, S. G.; Goodwin, C. J. Tetrahedron: Asymmetry 1994, 5, 203–206. doi:10.1016/S0957-4166(00)86173-X |
| 11. | Li, G.; Chang, H.-T.; Sharpless, K. B. Angew. Chem., Int. Ed. 1996, 35, 451–454. doi:10.1002/anie.199604511 |
| 12. | Chandrasekhar, S.; Mohapatra, S.; Yadav, J. S. Tetrahedron 1997, 8, 4089–4099. doi:10.1016/S0957-4166(97)00595-8 |
| 13. | Sugimura, H.; Miura, M.; Yamada, N. Tetrahedron: Asymmetry 1997, 8, 4089–4099. doi:10.1016/S0957-4166(97)00595-8 |
| 14. | Righi, G.; Chionne, A.; D’Achille, R.; Bonini, C. Tetrahedron: Asymmetry 1997, 8, 903–907. doi:10.1016/S0957-4166(97)00056-6 |
| 15. | Jeffords, C. W.; Mcnulty, J.; Lu, Z. H.; Wang, J. B. Helv. Chim. Acta 1996, 79, 1203–1216. doi:10.1002/hlca.19960790426 |
| 32. | Patil, N. T.; John, S.; Sabharwal, S. G.; Dhavale, D. D. Bioorg. Med. Chem. 2002, 10, 2155–2160. doi:10.1016/S0968-0896(02)00073-1 |
| 9. | Nicolau, K. C.; Dai, W.-M.; Guy, R. K. Angew. Chem., Int. Ed. 1994, 33, 15–44. doi:10.1002/anie.199400151 |
| 30. | Sato, K.-i.; Akai, S.; Sakuma, M.; Kojima, M.; Suzuki, K.-j. Tetrahedron Lett. 2003, 44, 4903–4907. doi:10.1016/S0040-4039(03)01098-0 |
| 5. | Umezawa, H.; Aoyagi, T.; Suda, H.; Hamada, M.; Takeuchi, T. J. Antibiot. 1976, 29, 97–99. doi:10.7164/antibiotics.29.97 |
| 6. | Nakamura, H.; Suda, H.; Takita, T.; Aoyagi, T.; Umezawa, H. J. Antibiot. 1976, 29, 102–103. doi:10.7164/antibiotics.29.102 |
| 7. | Sekizawa, R.; Iinuma, H.; Muraoka, Y.; Naganawa, H.; Kinoshita, N.; Nakamura, H.; Hamada, M.; Takeuchi, T.; Umezawa, K. J. Nat. Prod. 1996, 59, 232–236. doi:10.1021/np960067t |
| 8. | Tsuda, M.; Muraoka, Y.; Takeuchi, T. J. Antibiot. 1996, 49, 1031–1035. doi:10.7164/antibiotics.49.1031 |
| 24. | Dhavale, D. D.; Markad, S. D.; Karanjule, N. S.; Prakasha Reddy, J. J. Org. Chem. 2004, 69, 4760–4766. doi:10.1021/jo049509t |
| 25. | Karanjule, N. S.; Markad, S. D.; Dhavale, D. D. J. Org. Chem. 2006, 71, 6273–6276. doi:10.1021/jo060823s |
| 26. | Karanjule, N. S.; Markad, S. D.; Shinde, V. S.; Dhavale, D. D. J. Org. Chem. 2006, 71, 4667–4670. doi:10.1021/jo0601617 |
| 27. | Dhavale, D. D.; Ajish Kumar, K. S.; Chaudhari, V. D.; Sharma, T.; Sabharwal, S. G.; Prakasha, R. Org. Biomol. Chem. 2005, 3, 3720–3726. doi:10.1039/b509216g |
| 28. | Ajish Kumar, K. S.; Chaudhari, V. D.; Puranik, V. G.; Dhavale, D. D. Eur. J. Org. Chem. 2007, 4895–4901. doi:10.1002/ejoc.200700461 |
| 29. | Pawar, N. J.; Parihar, V.; Chavan, S.; Joshi, R.; Joshi, P. V.; Sabharwal, S. G.; Puranik, V. G.; Dhavale, D. D. J. Org. Chem. 2012, 77, 7873–7882. doi:10.1021/jo3009534 |
| 2. | Wyvratt, M. J.; Patchett, A. A. Med. Res. Rev. 1985, 5, 483–531. doi:10.1002/med.2610050405 |
| 3. | Moore, R. E.; Banarjee, S.; Bomemann, V.; Caplan, F. R.; Chen, J. L.; Corley, D. G.; Larsen, L. K.; Moore, B. S.; Patterson, G. M. L.; Paul, V. J.; Stewart, J. B.; Williams, D. E. Pure Appl. Chem. 1989, 61, 521–524. doi:10.1351/pac198961030521 |
| 4. | Carmichael W. W. Handbook of Natural Toxins; Tu, A. T.; Ed.; Marcel Dekker: New York, 1988; pp. 121 ff. |
| 30. | Sato, K.-i.; Akai, S.; Sakuma, M.; Kojima, M.; Suzuki, K.-j. Tetrahedron Lett. 2003, 44, 4903–4907. doi:10.1016/S0040-4039(03)01098-0 |
| 31. | Hanessian, S. Total Synthesis of Natural Products: The "Chiron" Approach; Pergamon Press: Oxford, 1983. |
| 19. | Tuch, A.; Saniere, M.; Merrer, Y. L.; Depezay, J.-C. Tetrahedron: Asymmetry 1996, 7, 2901–2909. doi:10.1016/0957-4166(96)00381-3 |
| 21. | Shirode, N. M.; Deshmukh, A. R. A. S. Tetrahedron 2006, 62, 4615–4621. doi:10.1016/j.tet.2006.01.082 |
| 33. | Bindra, J.; Grodski, A. J. Org. Chem. 1978, 43, 3240. doi:10.1021/jo00410a031 |
| 18. | Wee, A. G. H.; McLeod, D. D. J. Org. Chem. 2003, 68, 6268–6273. doi:10.1021/jo034334t |
| 22. | Kalamkar, N. B.; Kasture, V. M.; Dhavale, D. D. Tetrahedron Lett. 2010, 51, 6745–6747. doi:10.1016/j.tetlet.2010.10.086 |
| 23. | Kalamkar, N. B.; Kasture, V. M.; Dhavale, D. D. J. Org. Chem. 2008, 73, 3619–3622. doi:10.1021/jo702749r |
| 21. | Shirode, N. M.; Deshmukh, A. R. A. S. Tetrahedron 2006, 62, 4615–4621. doi:10.1016/j.tet.2006.01.082 |
| 17. | Gassa, F.; Contini, A.; Fontana, G.; Pellegrino, S.; Gelmi, M. L. J. Org. Chem. 2010, 75, 7099–7106. doi:10.1021/jo1011762 |
| 36. | Carlsen, P. J. H.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045 |
| 16. | Ha, H. J.; Ahn, Y. G.; Woo, J. S.; Lee, G. S.; Lee, W. K. Bull. Chem. Soc. Jpn. 2001, 74, 1667–1672. doi:10.1246/bcsj.74.1667 |
| 20. | Bergmeier, S. C.; Stanchina, D. M. J. Org. Chem. 1999, 64, 2852–2859. doi:10.1021/jo9823893 |
| 18. | Wee, A. G. H.; McLeod, D. D. J. Org. Chem. 2003, 68, 6268–6273. doi:10.1021/jo034334t |
© 2014 Rohokale and Dhavale; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)