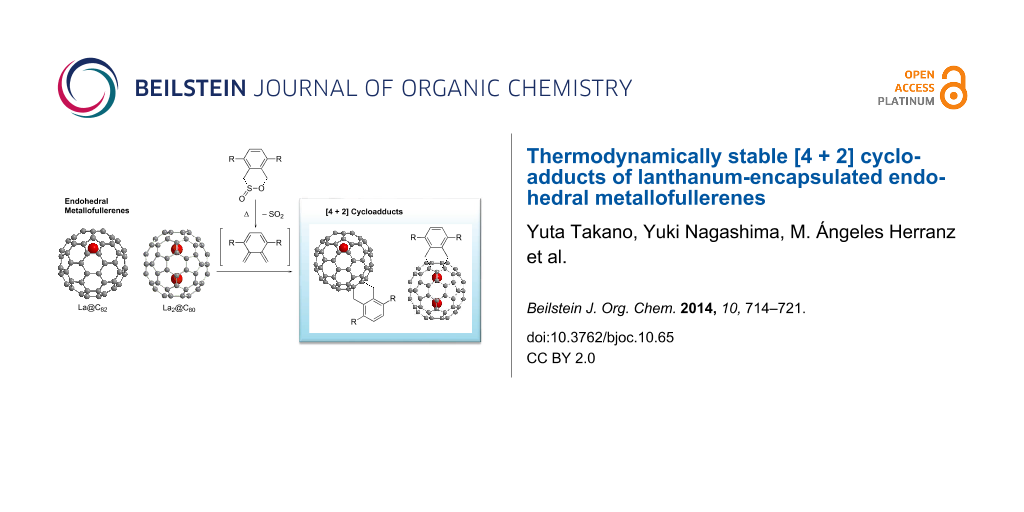
Abstract
The [4 + 2] cycloaddition of o-quinodimethanes, generated in situ from the sultine 4,5-benzo-3,6-dihydro-1,2-oxathiin 2-oxide and its derivative, to La metal-encapsulated fullerenes, La2@C80 or La@C82, afforded the novel derivatives of endohedral metallofullerenes (3a,b, 4a,b and 5b). Molecular structures of the resulting compounds were elucidated using spectroscopic methods such as MALDI–TOF mass, optical absorption, and NMR spectroscopy. The [4 + 2] adducts of La2@C80 (3a,b, and 4a,b) and La@C82 (5b), respectively, retain diamagnetic and paramagnetic properties, as confirmed by EPR spectroscopy. Dynamic NMR measurements of 4a at various temperatures demonstrated the boat-to-boat inversions of the addend. In addition, 5b revealed remarkable thermal stability in comparison with the reported [4 + 2] cycloadduct of pentamethylcyclopentadiene and La@C82 (6). These findings demonstrate the utility of sultines to afford thermodynamically stable endohedral metallofullerene derivatives for the use in material science.

Graphical Abstract
Introduction
Endohedral metallofullerenes (EMFs) are a family of nanocarbons, which encapsulated one or more metal atoms inside a hollow carbon cage [1-4]. The encapsulation results in the electron transfer from metal atoms to the fullerene cage, which leads to unique electronic, magnetic, and chemical properties for EMFs that cannot be expected for empty fullerenes. Due to the numerous electronic properties EMFs are anticipated as promising materials in various fields such as chemistry, biology, and material science.
Among various kinds of EMFs, those encapsulating La atoms are especially attractive molecules because of their electronic and magnetic characteristics. As a result of the three electron transfer per La atom to the fullerene cage, the fullerenes simultaneously possess a low ionizing potential and a high electron affinity [1,2]. For mono-La endohedral fullerenes such as La@C82, the electron transfer results in paramagnetism of the fullerene cage [5]. The di-La endohedral fullerenes such as La2@C80 show diamagnetism [6]. This feature leads to remarkable differences in chemical and electronic properties between these two classes of EMFs.
Chemical functionalization of fullerenes enhances molecular properties and possible applications of fullerenes [1,4]. The [4 + 2] cycloaddition reaction is a useful chemical modification method because it enables to introduce a variety of addends and/or the combination of different functionalities on the fullerene [7]. Regarding the [4 + 2] cycloaddition of EMFs, however, no report is available for endohedral di-metallofullerenes, with the exception of azafullerene [8]. A limited number of reports describe other EMFs [4,9,10]. Moreover, the only precedent of [4 + 2] cycloadducts of the fullerenes which have an open-shell electronic structure of the cage, e.g., La@C82, are thermodynamically unstable and show retro-cycloaddition reactions [10,11]. In addition, boat-to-boat inversion of the addend of the cycloadducts of EMFs has not been well-studied to date; the investigation of this interconversion serves to demonstrate the existence of a dynamic process in the molecules and is regarded as one index of the bonding energy of the addition position of the fullerene and the addend.
Among various precursors to afford [4 + 2] cycloadducts of fullerenes, the sultine 4,5-benzo-3,6-dihydro-1,2-oxathiin 2-oxide and its derivatives are useful to afford thermodynamically stable compounds because thermolysis of sultine affords highly reactive o-quinodimethanes by extrusion of sulfur dioxide without production of any organic or inorganic byproduct [12,13].
Here, we present the first chemical derivatization of La2@C80 and La@C82 by [4 + 2] cycloaddition using sultines 1a,b. The resulting products 3a,b, 4a,b and 5b were characterized and their thermodynamic properties were investigated.
Results and Discussion
Synthesis and characterization of La2@C80 cycloadducts
o-Quinodimethanes 2a and 2b were generated in situ by thermolysis of the corresponding sultines 1a and 1b in toluene at 80 °C (Scheme 1). The highly reactive intermediates are trapped efficiently by La2@C80, which acts as a dienophile to form the cycloadducts (3a,b and 4a,b). The reactions were traced using HPLC analyses (Figure 1), and formation of the resulting [4 + 2] adducts was confirmed by matrix-assisted laser desorption ionization (MALDI) TOF mass spectrometry (Figure 2), which shows the molecular ion peaks for the corresponding compounds. Isolation of 3b and 4b were achieved by one-step HPLC separation using a Buckyprep column, although this purification method was not applicable for 3a and 4a (vide infra).
![[1860-5397-10-65-i1]](/bjoc/content/inline/1860-5397-10-65-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of [4 + 2] adducts of La2@C80.
Scheme 1: Synthesis of [4 + 2] adducts of La2@C80.
![[1860-5397-10-65-1]](/bjoc/content/figures/1860-5397-10-65-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: HPLC profiles of the reaction solutions (black) before and (red) after the reaction of La2@C80 and (a) 1a and (b) 1b, respectively. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); eluent, toluene; flow rate, 1.0 mL/min; wavelength: 330 nm; rt.
Figure 1: HPLC profiles of the reaction solutions (black) before and (red) after the reaction of La2@C80 and ...
![[1860-5397-10-65-2]](/bjoc/content/figures/1860-5397-10-65-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: MALDI–TOF mass (negative mode) spectra of (a) 3b and (b) 4b, using 1,1,4,4-tetraphenyl-1,3-butadiene as matrix.
Figure 2: MALDI–TOF mass (negative mode) spectra of (a) 3b and (b) 4b, using 1,1,4,4-tetraphenyl-1,3-butadien...
Because only two types of C=C bonds are available in La2@C80, which has Ih symmetry, only two site-isomers namely [6,6]- and [5,6]-isomers, are allowed to be formed by cycloaddition reactions. (Please note that “site-isomer” refers to an isomer of the adducts which has the same fullerene and addend but different addition position – a classification proposed recently for fullerene’s chemistry by Martin et al. [14]). Therefore, 3b and 4b are concluded to be the site-isomers which were afforded by the reaction as a result of using highly reactive o-quinodimethane.
The UV–vis spectra of 3b and 4b partially provide information related to their molecular structures. The spectra were firstly recorded using a diode-array detector of the HPLC apparatus (Figure 3). The spectrum of 3b shows the specific absorption band around 700 nm, which strongly suggests the electric nature of a [6,6]-closed adduct of the La2@C80 derivatives [15], because the absorption spectra of fullerenes and their derivatives are mainly attributable to π–π* transitions, which reflects the distinctive fingerprints of the π-electron system topology of the fullerene cage. Similarly, the characteristic spectrum of 4b suggests its [5,6]-closed structure.
![[1860-5397-10-65-3]](/bjoc/content/figures/1860-5397-10-65-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis/near-IR absorption spectra of 3b and 4b recorded by the diode array detector of the HPLC apparatus.
Figure 3: UV–vis/near-IR absorption spectra of 3b and 4b recorded by the diode array detector of the HPLC app...
A different approach was taken to purify 3a and 4a because of the similar retention time of these compounds in HPLC. The mixture of 3a and 4a was first separated from the unreacted starting materials and byproducts through one-step HPLC separation. The MALDI–TOF mass spectra of 3a and 4a showed single peaks attributed to the molecular ion peak of the target molecule, La2@C80C2H4C6H4, at 1342 m/z (Figure 4). The existence of both [6,6]- and [5,6]-isomers in the mixture was indicated by the 1H NMR spectrum recorded at 248 K (Figure 5b), which cannot be expected from a single regioisomer. The [6,6]-adduct should show only two sets of AB quartets. The [5,6]-adduct should demonstrate one or two AB quartets based on its Cs molecular symmetry. Consequently, the spectrum containing more than two AB quartets indicates the existence of the both site-isomers.
![[1860-5397-10-65-4]](/bjoc/content/figures/1860-5397-10-65-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: MALDI–TOF mass spectrum (negative mode) of the reaction mixture from La2@C80 and 1a, using 1,1,4,4-tetraphenyl-1,3-butadiene as matrix.
Figure 4: MALDI–TOF mass spectrum (negative mode) of the reaction mixture from La2@C80 and 1a, using 1,1,4,4-...
![[1860-5397-10-65-5]](/bjoc/content/figures/1860-5397-10-65-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 1H NMR spectra of (a) the mixture of 3a and 4a in C2D2Cl4 at 248 K, and (b) isolated 4a at 230 K, recorded in 300 MHz.
Figure 5: 1H NMR spectra of (a) the mixture of 3a and 4a in C2D2Cl4 at 248 K, and (b) isolated 4a at 230 K, r...
Further isolation of 4a and 3b was respectively accomplished using a combination of heating and HPLC separation. When the powdery mixture of 3a and 4a was heated to a temperature of 250 °C, selective decomposition of 3a was observed (Figure 6). Since the peak of pristine La@C82 at ca. 30 min is observed after heating (see Figure 6b), most probably the detachment of the addend is taking place and a thermal isomerization thereafter. After HPLC purification of the crude reaction mixture, the number of the 1H NMR signals attributed to the methylene protons was reduced (Figure 5b). Furthermore, the 1H NMR spectrum of 4a at 230 K unambiguously shows the existence of a single regioisomer, a [5,6]-adduct, which has Cs symmetry showing one AB quartet of the one set of equivalent methylene protons. The existence of the [6,6]-adduct is excluded because the adduct must show at least two AB quartets based on its molecular symmetry.
![[1860-5397-10-65-6]](/bjoc/content/figures/1860-5397-10-65-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: HPLC profiles of the mixture of 3a and 4a, (a) after heating in refluxing 1,2-dichlorobenzene and (b) after heating at 250 °C. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); eluent, toluene; flow rate, 1.0 mL/min; wavelength, 330 nm; rt.
Figure 6: HPLC profiles of the mixture of 3a and 4a, (a) after heating in refluxing 1,2-dichlorobenzene and (...
In the case of purifying 3b, selective decomposition of 4b was observed at much lower temperature than that of 3a and 4a, after refluxing the mixture of 3b and 4b in toluene (Figure 7). This phenomenon is rationalized by the decomposition of the addend itself, because no pristine La@C82 was detected after the heating in contrast to the case of 3a and 4a (vide supra). Therefore, it is concluded that the addend of 4b containing methoxy groups is thermally less stable than the addend of 3a and 4a. The following HPLC purification afforded isolated 3b. This result suggests that 3b is more stable against heating than 4b.
![[1860-5397-10-65-7]](/bjoc/content/figures/1860-5397-10-65-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: HPLC profiles of the reaction mixture of 3b and 4b, (black) before and (red) after heating in refluxing toluene. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); eluent, toluene; flow rate, 1.0 mL/min; wavelength, 330 nm; rt.
Figure 7: HPLC profiles of the reaction mixture of 3b and 4b, (black) before and (red) after heating in reflu...
The UV–vis spectra of purified 4a and 3b were recorded using a spectrophotometer (UV-3150; Shimadzu Corp.) instead of the HPLC apparatus (Figure 8). The spectra reveal that 4a shows no clear absorption peaks in the measuring range, and demonstrates a similar spectrum to those of [5,6]-adducts of La2@C80 [15]. This result shows good agreement with the 1H NMR spectrum, which indicates that 4a is a [5,6]-adduct. However, 3b shows a specific absorption band around 700 nm as in the spectrum recorded by the HPLC system shown in Figure 3, indicating its [6,6]-structure.
![[1860-5397-10-65-8]](/bjoc/content/figures/1860-5397-10-65-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: UV–vis/near-IR absorption spectra of 3b and 4a in toluene.
Figure 8: UV–vis/near-IR absorption spectra of 3b and 4a in toluene.
Temperature-dependent dynamics of the [4 + 2] adducts of endohedral metallofullerenes were studied for 4a by dynamic 1H NMR measurements. Although 4a did not show clear peaks at 290 K, distinct peaks were observed when the temperature was sufficiently lower or high enough distant from the coalescence temperature (Tc) (Figure 9). This fact suggests that the boat-to-boat inversions of 4a between the pentagon side and the hexagon side are sufficiently slow or fast to allow their observation in an NMR time scale. The signals from the AB-quartet of 4a coalesce at 290 K (= Tc) indicate a dynamic process, which is attributed to the boat-to-boat interconversion of the cyclohexane ring of the addend similarly to the related carbocyclic analogues of C60 adducts [12].
![[1860-5397-10-65-9]](/bjoc/content/figures/1860-5397-10-65-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Temperature-dependent 1H NMR spectra of 4a in C2D2Cl4 (left) at 300 MHz, and (right) at 500 MHz for precise analysis.
Figure 9: Temperature-dependent 1H NMR spectra of 4a in C2D2Cl4 (left) at 300 MHz, and (right) at 500 MHz for...
Synthesis and characterization of La@C82 cycloadducts
Thermal reactions of 1b and La@C82 afforded the [4 + 2] adduct 5b (Scheme 2). 5b was separated from the unreacted starting materials and byproducts through a one-step HPLC procedure (Figure 10). The MALDI–TOF mass spectrum of 5b shows the peak attributed to the molecular ion peak (Figure 11).
![[1860-5397-10-65-i2]](/bjoc/content/inline/1860-5397-10-65-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of [4 + 2] adducts of La@C82.
Scheme 2: Synthesis of [4 + 2] adducts of La@C82.
![[1860-5397-10-65-10]](/bjoc/content/figures/1860-5397-10-65-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: HPLC profiles of the reaction mixture for 5b. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); eluent, toluene; flow rate, 1.0 mL/min; wavelength: 330 nm; 40 °C.
Figure 10: HPLC profiles of the reaction mixture for 5b. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); el...
![[1860-5397-10-65-11]](/bjoc/content/figures/1860-5397-10-65-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: MALDI–TOF mass spectra (negative mode) of 5b, using 1,1,4,4-tetraphenyl-1,3-butadiene as matrix.
Figure 11: MALDI–TOF mass spectra (negative mode) of 5b, using 1,1,4,4-tetraphenyl-1,3-butadiene as matrix.
The electron spin resonance (ESR) spectrum of 5b showed a unique octet signal (see Supporting Information File 1), indicating the paramagnetic property of 5b as well as pristine La@C82. This result also indicates that the cycloaddition of the o-quinodimethane does not lead to a remarkable change in the electronic properties of the fullerene. This fact is supported by the vis–NIR absorption spectrum of 5b, which retains the specific absorption bands of the pristine La@C82 (Figure 12). Broadening of the absorption bands is also observed, which is expected to be caused by the reduction of the molecular symmetry from C2v to C1 (vide infra).
![[1860-5397-10-65-12]](/bjoc/content/figures/1860-5397-10-65-12.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Vis–near-IR spectra of 5b, 6 and La@C82 in CS2.
Figure 12: Vis–near-IR spectra of 5b, 6 and La@C82 in CS2.
Further characterization of the molecular structure was performed using NMR measurements. Because 5b has an open-shell electronic structure as well as pristine La@C82, 5b was reduced electrochemically by one electron using bulk potential electrolysis for the NMR measurements. 1H NMR spectrum of the resulting anionic 5b ([5b]−) clearly illustrates the characteristic signals from the addend (Figure 13). Signals of the methylene protons appear as a sharp AB system at 4.55, 4.30, 2.94, and 2.82 ppm. The 13C NMR spectrum demonstrates the total sum of 82 signals from the carbon cage (Figure 14), indicating C1 molecular symmetry for [5b]−. Signals at 58.2 and 56.0 ppm are attributed to the sp3 carbons of the addition position of the addend.
![[1860-5397-10-65-13]](/bjoc/content/figures/1860-5397-10-65-13.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: 1H NMR spectrum of [5b]− in acetone-d6/CS2 (3/1 = v/v) at 223 K.
Figure 13: 1H NMR spectrum of [5b]− in acetone-d6/CS2 (3/1 = v/v) at 223 K.
![[1860-5397-10-65-14]](/bjoc/content/figures/1860-5397-10-65-14.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: 13C NMR spectrum of [5b]− in acetone-d6/CS2 (3/1 = v/v).
Figure 14: 13C NMR spectrum of [5b]− in acetone-d6/CS2 (3/1 = v/v).
The vis–NIR absorption spectra provide information related to the molecular structure of 5b (Figure 12). The resemblance between the spectra of 5b and that of the previously reported [4 + 2] cycloadduct of La@C82 (6: La@C82Cp*) [11] in the vis–NIR region imply the isostructural characteristics of the respective compounds, which show the same addition pattern of the substituents. Although the structure of 5b was not elucidated using X-ray crystallographic analysis, synthetic precedents with absorption spectral data and theoretical calculations [16] strongly suggest that the most feasible addition site of the addend is that indicated in Scheme 2.
The thermodynamic stability of 5b was evaluated by thermal heating. When a toluene solution of 5b was let to stand at 30 °C, no decomposition was observed, whereas 6 showed decomposition and generation of pristine La@C82 (Figure 15). This result shows that using sultines, which generate reactive o-quinodimethanes and which afford cycloadducts, is an effective means to afford thermodynamically stable [4 + 2] adducts of La@C82.
![[1860-5397-10-65-15]](/bjoc/content/figures/1860-5397-10-65-15.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: HPLC profiles for comparison of the thermal stabilities of (a) 5b and (b) 6 at 30 °C. Conditions: column, Buckyprep (Ø 4.6 mm × 250 mm); temperature, 40 °C.
Figure 15: HPLC profiles for comparison of the thermal stabilities of (a) 5b and (b) 6 at 30 °C. Conditions: c...
Conclusion
In summary, novel cycloadducts of La2@C80 and La@C82 were synthesized efficiently by a [4 + 2] cycloaddition reaction using sultines as a precursor of reactive o-quinodimethanes. Isolation of 3b and 4a, respectively, was achieved by the selective thermal decomposition of unstable isomers. The thermal stability of 5b was also evaluated in comparison with 6, and 5b shows remarkable thermal stability. The molecular structures of the resulting compounds were characterized by spectroscopic analyses. Dynamic 1H NMR spectroscopic investigations of 4a reveal temperature-dependent changes related to the conformational changes in the cyclohexane moiety generated upon reaction. The use of sultines for chemical modification of endohedral metallofullerenes has proved to be of general scope, being particularly useful to prepare thermally stable [4 + 2] cycloadducts.
Supporting Information
Supporting information features detailed experimental procedures and spectral data for the compounds.
| Supporting Information File 1: Descriptions on the synthesis and analyses of the compounds. | ||
| Format: PDF | Size: 651.1 KB | Download |
Acknowledgements
This work is supported by a Grant-in-Aid for Scientific Research on Innovative Areas (20108001, "pi-Space"), a Grant-in-Aid for Scientific Research (A) (202455006), (B) (24350019) and Young Scientists (B) (25810098) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, The Strategic Japanese-Spanish Cooperative Program funded by JST and MINECO (Projects PLE-2009-0039 and PIB2010JP-00196), and by the European Research Council ERC-2012-ADG_20120216 (Chirallcarbon). The iCEMS is supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan. N. M. thanks to Alexander van Humboldt Foundation.
References
-
Akasaka, T.; Wudl, F.; Nagase, S., Eds. Chemistry of Nanocarbons; Wiley: Chichester, 2010.
Return to citation in text: [1] [2] [3] -
Chaur, M. N.; Melin, F.; Ortiz, A. L.; Echegoyen, L. Angew. Chem., Int. Ed. 2009, 48, 7514–7538. doi:10.1002/anie.200901746
Return to citation in text: [1] [2] -
Rudolf, M.; Wolfrum, S.; Guldi, D. M.; Feng, L.; Tsuchiya, T.; Akasaka, T.; Echegoyen, L. Chem.–Eur. J. 2012, 18, 5136–5148. doi:10.1002/chem.201102844
Return to citation in text: [1] -
Popov, A. A.; Yang, S.; Dunsch, L. Chem. Rev. 2013, 113, 5989–6113. doi:10.1021/cr300297r
Return to citation in text: [1] [2] [3] -
Nagase, S.; Kobayashi, K. J. Chem. Soc., Chem. Commun. 1994, 1837–1838. doi:10.1039/c39940001837
Return to citation in text: [1] -
Kobayashi, K.; Nagase, S.; Akasaka, T. Chem. Phys. Lett. 1996, 261, 502–506. doi:10.1016/0009-2614(96)01026-3
Return to citation in text: [1] -
Hirsch, A.; Bettreich, M. Fullerenes, Chemistry and Reaction; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Fu, W.; Zhang, J.; Fuhrer, T.; Champion, H.; Furukawa, K.; Kato, T.; Mahaney, J. E.; Burke, B. G.; Williams, K. A.; Walker, K.; Dixon, C.; Ge, J.; Shu, C.; Harich, K.; Dorn, H. C. J. Am. Chem. Soc. 2011, 133, 9741–9750. doi:10.1021/ja202011u
Return to citation in text: [1] -
Lee, H. M.; Olmstead, M. M.; Iezzi, E.; Duchamp, J. C.; Dorn, H. C.; Balch, A. L. J. Am. Chem. Soc. 2002, 124, 3494–3495. doi:10.1021/ja020065x
Return to citation in text: [1] -
Maeda, Y.; Miyashita, J.; Hasagawa, T.; Wakahara, T.; Tsuchiya, T.; Nakahodo, T.; Akasaka, T.; Mizorogi, N.; Kobayashi, K.; Nagase, S.; Kato, T.; Ban, N.; Nakajima, H.; Watanabe, Y. J. Am. Chem. Soc. 2005, 127, 12190–12191. doi:10.1021/ja053983e
Return to citation in text: [1] [2] -
Maeda, Y.; Sato, S.; Inada, K.; Nikawa, H.; Yamada, M.; Mizorogi, N.; Hasegawa, T.; Tsuchiya, T.; Akasaka, T.; Kato, T.; Slanina, Z.; Nagase, S. Chem.–Eur. J. 2010, 16, 2193–2197. doi:10.1002/chem.200902512
Return to citation in text: [1] [2] -
Illescas, B. M.; Martín, N.; Seoane, C.; Ortí, E.; Viruela, P. M.; Viruela, R.; de la Hoz, A. J. Org. Chem. 1997, 62, 7585–7591. doi:10.1021/jo9706436
Return to citation in text: [1] [2] -
Segura, J. L.; Martín, N. Chem. Rev. 1999, 99, 3199–3246. doi:10.1021/cr990011e
Return to citation in text: [1] -
Maroto, E. E.; de Cózar, A.; Filippone, S.; Martin-Domenech, A.; Suarez, M.; Cossío, F. P.; Martín, N. Angew. Chem., Int. Ed. 2011, 50, 6060–6064. doi:10.1002/anie.201101246
Return to citation in text: [1] -
Yamada, M.; Wakahara, T.; Nakahodo, T.; Tsuchiya, T.; Maeda, Y.; Akasaka, T.; Yoza, K.; Horn, E.; Mizorogi, N.; Nagase, S. J. Am. Chem. Soc. 2006, 128, 1402–1403. doi:10.1021/ja056560l
Return to citation in text: [1] [2] -
Takano, Y.; Obuchi, S.; Mizorogi, N.; García, R.; Herranz, M. Á.; Rudolf, M.; Wolfrum, S.; Guldi, D. M.; Martín, N.; Nagase, S.; Akasaka, T. J. Am. Chem. Soc. 2012, 134, 16103–16106. doi:10.1021/ja3055386
Return to citation in text: [1]
| 1. | Akasaka, T.; Wudl, F.; Nagase, S., Eds. Chemistry of Nanocarbons; Wiley: Chichester, 2010. |
| 2. | Chaur, M. N.; Melin, F.; Ortiz, A. L.; Echegoyen, L. Angew. Chem., Int. Ed. 2009, 48, 7514–7538. doi:10.1002/anie.200901746 |
| 3. | Rudolf, M.; Wolfrum, S.; Guldi, D. M.; Feng, L.; Tsuchiya, T.; Akasaka, T.; Echegoyen, L. Chem.–Eur. J. 2012, 18, 5136–5148. doi:10.1002/chem.201102844 |
| 4. | Popov, A. A.; Yang, S.; Dunsch, L. Chem. Rev. 2013, 113, 5989–6113. doi:10.1021/cr300297r |
| 1. | Akasaka, T.; Wudl, F.; Nagase, S., Eds. Chemistry of Nanocarbons; Wiley: Chichester, 2010. |
| 4. | Popov, A. A.; Yang, S.; Dunsch, L. Chem. Rev. 2013, 113, 5989–6113. doi:10.1021/cr300297r |
| 11. | Maeda, Y.; Sato, S.; Inada, K.; Nikawa, H.; Yamada, M.; Mizorogi, N.; Hasegawa, T.; Tsuchiya, T.; Akasaka, T.; Kato, T.; Slanina, Z.; Nagase, S. Chem.–Eur. J. 2010, 16, 2193–2197. doi:10.1002/chem.200902512 |
| 6. | Kobayashi, K.; Nagase, S.; Akasaka, T. Chem. Phys. Lett. 1996, 261, 502–506. doi:10.1016/0009-2614(96)01026-3 |
| 16. | Takano, Y.; Obuchi, S.; Mizorogi, N.; García, R.; Herranz, M. Á.; Rudolf, M.; Wolfrum, S.; Guldi, D. M.; Martín, N.; Nagase, S.; Akasaka, T. J. Am. Chem. Soc. 2012, 134, 16103–16106. doi:10.1021/ja3055386 |
| 5. | Nagase, S.; Kobayashi, K. J. Chem. Soc., Chem. Commun. 1994, 1837–1838. doi:10.1039/c39940001837 |
| 15. | Yamada, M.; Wakahara, T.; Nakahodo, T.; Tsuchiya, T.; Maeda, Y.; Akasaka, T.; Yoza, K.; Horn, E.; Mizorogi, N.; Nagase, S. J. Am. Chem. Soc. 2006, 128, 1402–1403. doi:10.1021/ja056560l |
| 1. | Akasaka, T.; Wudl, F.; Nagase, S., Eds. Chemistry of Nanocarbons; Wiley: Chichester, 2010. |
| 2. | Chaur, M. N.; Melin, F.; Ortiz, A. L.; Echegoyen, L. Angew. Chem., Int. Ed. 2009, 48, 7514–7538. doi:10.1002/anie.200901746 |
| 12. | Illescas, B. M.; Martín, N.; Seoane, C.; Ortí, E.; Viruela, P. M.; Viruela, R.; de la Hoz, A. J. Org. Chem. 1997, 62, 7585–7591. doi:10.1021/jo9706436 |
| 10. | Maeda, Y.; Miyashita, J.; Hasagawa, T.; Wakahara, T.; Tsuchiya, T.; Nakahodo, T.; Akasaka, T.; Mizorogi, N.; Kobayashi, K.; Nagase, S.; Kato, T.; Ban, N.; Nakajima, H.; Watanabe, Y. J. Am. Chem. Soc. 2005, 127, 12190–12191. doi:10.1021/ja053983e |
| 11. | Maeda, Y.; Sato, S.; Inada, K.; Nikawa, H.; Yamada, M.; Mizorogi, N.; Hasegawa, T.; Tsuchiya, T.; Akasaka, T.; Kato, T.; Slanina, Z.; Nagase, S. Chem.–Eur. J. 2010, 16, 2193–2197. doi:10.1002/chem.200902512 |
| 14. | Maroto, E. E.; de Cózar, A.; Filippone, S.; Martin-Domenech, A.; Suarez, M.; Cossío, F. P.; Martín, N. Angew. Chem., Int. Ed. 2011, 50, 6060–6064. doi:10.1002/anie.201101246 |
| 4. | Popov, A. A.; Yang, S.; Dunsch, L. Chem. Rev. 2013, 113, 5989–6113. doi:10.1021/cr300297r |
| 9. | Lee, H. M.; Olmstead, M. M.; Iezzi, E.; Duchamp, J. C.; Dorn, H. C.; Balch, A. L. J. Am. Chem. Soc. 2002, 124, 3494–3495. doi:10.1021/ja020065x |
| 10. | Maeda, Y.; Miyashita, J.; Hasagawa, T.; Wakahara, T.; Tsuchiya, T.; Nakahodo, T.; Akasaka, T.; Mizorogi, N.; Kobayashi, K.; Nagase, S.; Kato, T.; Ban, N.; Nakajima, H.; Watanabe, Y. J. Am. Chem. Soc. 2005, 127, 12190–12191. doi:10.1021/ja053983e |
| 15. | Yamada, M.; Wakahara, T.; Nakahodo, T.; Tsuchiya, T.; Maeda, Y.; Akasaka, T.; Yoza, K.; Horn, E.; Mizorogi, N.; Nagase, S. J. Am. Chem. Soc. 2006, 128, 1402–1403. doi:10.1021/ja056560l |
| 8. | Fu, W.; Zhang, J.; Fuhrer, T.; Champion, H.; Furukawa, K.; Kato, T.; Mahaney, J. E.; Burke, B. G.; Williams, K. A.; Walker, K.; Dixon, C.; Ge, J.; Shu, C.; Harich, K.; Dorn, H. C. J. Am. Chem. Soc. 2011, 133, 9741–9750. doi:10.1021/ja202011u |
| 7. | Hirsch, A.; Bettreich, M. Fullerenes, Chemistry and Reaction; Wiley-VCH: Weinheim, Germany, 2005. |
| 12. | Illescas, B. M.; Martín, N.; Seoane, C.; Ortí, E.; Viruela, P. M.; Viruela, R.; de la Hoz, A. J. Org. Chem. 1997, 62, 7585–7591. doi:10.1021/jo9706436 |
| 13. | Segura, J. L.; Martín, N. Chem. Rev. 1999, 99, 3199–3246. doi:10.1021/cr990011e |
© 2014 Takano et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)