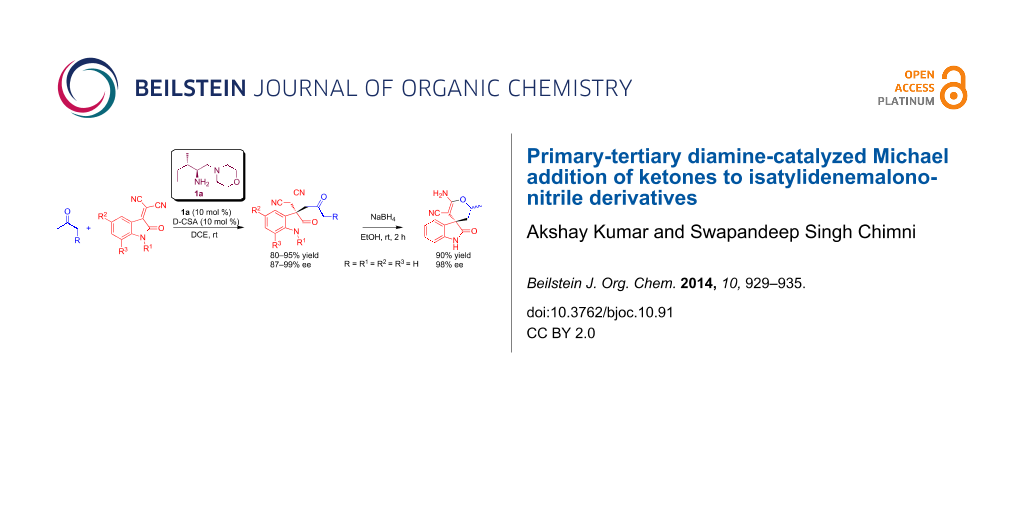
Abstract
Simple primary-tertiary diamines easily derived from natural primary amino acids were used to catalyze the Michael addition of ketones with isatylidenemalononitrile derivatives. Diamine 1a in combination with D-CSA as an additive provided Michael adducts in high yield (up to 94%) and excellent enantioselectivity (up to 99%). The catalyst 1a was successfully used to catalyze the three-component version of the reaction by a domino Knoevenagel–Michael sequence. The Michael adduct 4a was transformed into spirooxindole 6 by a reduction with sodium borohydride in a highly enantioselective manner.

Graphical Abstract
Introduction
The Michael reaction is one of the fundamental carbon–carbon bond forming reactions in organic synthesis, since a plethora of carbon nucleophiles and activated olefins could be expected to give versatile arrangements [1-8]. Among the various Michael acceptors, the oxindole-based Michael acceptors (Figure 1) are considered as valuable electrophiles for catalytic Michael reactions, as these provide a viable approach to procure 3,3'-disubstituted oxindole frameworks [9-16]. The oxindole framework bearing a tetra-substituted carbon stereocenter at C-3 is a privileged heterocyclic motif that is present in a large variety of bioactive natural products and a series of pharmaceutically active compounds [17]. In recent years, isatins derived Michael acceptors, isatylidenemalononitriles, have attracted considerable attention as novel substrates for the enantioselective synthesis of 3,3'-disubstituted oxindoles [18-22]. The organocatalytic Michael addition of ketones to isatylidenemalononitriles via enamine-catalysis has emerged as a useful tool for the synthesis of 3,3'-disubstituted oxindoles, which serve as important precursors to procure various structurally diverse spirooxindoles [23,24].
![[1860-5397-10-91-1]](/bjoc/content/figures/1860-5397-10-91-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Oxindole based Michael acceptors.
Figure 1: Oxindole based Michael acceptors.
Over the years, many chiral organocatalysts have been developed and explored for Michael reactions [1-8]. Recently, aminocatalysts – in particular those bearing a primary amine moiety – have been found to catalyze a variety of carbon–carbon bond-forming reactions [25-30]. Small peptides derived from acyclic amino acids, primary-secondary diamines, Cinchona-based primary amines, and thioureas with a primary amine functionality etc., have found many successful applications in Michael addition reactions via an iminium–enamine catalysis [31-37]. A few applications of primary-tertiary diamine in aldol reactions have been published [39-43]. To the best of our knowledge, however, the catalytic potential of amino acids derived primary-tertiary diamine organocatalysts for Michael reaction via enamine activation has not been investigated so far [38]. With readily available and inexpensive natural amino acids as a chiral source, we developed very simple primary-tertiary diamine organocatalysts (Figure 2) for asymmetric aldol reactions [44,45]. We describe herein that a similar catalyst system could also efficiently catalyze the asymmetric Michael addition reaction between ketones 2 and isatylidenemalononitrile derivatives 3 to procure highly functionalized 3,3'-disubstituted oxindoles 4 which could easily be transformed into spirooxindoles.
![[1860-5397-10-91-2]](/bjoc/content/figures/1860-5397-10-91-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Primary-tertiary diamine organocatalysts.
Figure 2: Primary-tertiary diamine organocatalysts.
Results and Discussion
Initially, the Michael addition of acetone (2a) to isatylidenemalononitrile (3a) catalyzed by chiral diamine 1a (10 mol %) with trichloroacetic acid (10 mol %) as an additive under mild conditions at room temperature was investigated (Scheme 1).
![[1860-5397-10-91-i1]](/bjoc/content/inline/1860-5397-10-91-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diamine catalyzed Michael addition of acetone to isatylidenemalononitrile.
Scheme 1: Diamine catalyzed Michael addition of acetone to isatylidenemalononitrile.
The Michael adduct 4a was isolated in 95% yield and 69% ee (Table 1, entry 1). Encouraged by the outcome of the preliminary reaction, we optimized the reaction conditions by studying the effect of different solvents and their amount as well as the effect of acid additives on this transformation (Tables 1–3). In our previous studies [10], the diamine 1a afforded the best result for aldol reactions with water as a solvent. Consequently, the above transformation was performed with water as a solvent. The Michael adduct 4a was obtained in good yield of 87% and a moderate enantioselectivity of 55% ee (Table 1, entry 2). It was planned to study the effect of different organic solvents on the stereochemical outcome of this reaction. Interestingly, the reaction of 2a with 3a performed in tetrahydrofuran (THF) gave Michael adduct 4a in good yield of 91% and a higher enantioselectivity of 84% ee (Table 1, entry 3). Other etheral solvents, such as 1,4-dioxane and methyl tert-butyl ether (MTBE), provided Michael adduct 4a in 89% and 90% yield and 89% ee and 87% ee, respectively (Table 1, entries 4 and 5). In toluene, 4a was obtained in 89% yield and 90% ee (Table 1, entry 6). The chlorinated solvents such as dichloromethane, chloroform and 1,2-dichloroethane (DCE) gave 4a in 90%, 89% and 91% yield and 90% ee, 88% ee and 91% ee, respectively (Table 1, entries 7–9). The utilization of methanol as a solvent led to the isolation of Michael adduct 4a in good yield but with a low enantioselectivity of 12% ee (Table 1, entry 11). Dimethylformamide was also found to be an inferior solvent for this reaction (Table 1, entry 12). Thus, 1,2-dichloroethane emerged as the solvent of choice for this transformation and was used for all further optimization studies (Table 1, entry 9).
Table 1: Solvent screening the Michael addition of acetone (2a) to isatylidenemalononitrile (3a) catalyzed by 1a TCA.a
| Entry | Solvent | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | – | 6 | 95 | 69 |
| 2 | water | 48 | 87 | 55 |
| 3 | THF | 30 | 91 | 84 |
| 4 | dioxane | 30 | 89 | 89 |
| 5 | MTBE | 30 | 90 | 87 |
| 6 | toluene | 36 | 89 | 90 |
| 7 | DCM | 36 | 90 | 90 |
| 8 | CHCl3 | 36 | 89 | 88 |
| 9 | ClCH2CH2Cl | 36 | 91 | 91 |
| 10 | ethyl acetate | 36 | 90 | 87 |
| 11 | CH3OH | 40 | 88 | 12 |
| 12 | DMF | 40 | 82 | 20 |
aReaction conditions: 1.5 mmol of acetone (2a), 0.125 mmol of isatylidenemalononitrile (3a), 0.25 mL of solvent, 10 mol % of catalyst 1a, 10 mol % of TCA, at 25 °C. bIsolated yield determined after chromatographic purification. cEnantiomeric excess determined by chiral HPLC.
In order to see the effect of the reaction concentration, the amount of 1,2-dichloroethane was varied (Table 2). The use of 0.5 mL of 1,2-dichloroethane afforded 4a in good yield of 93% and enantioselectivity of 91% ee (Table 2, entry 2). The reaction carried out with 0.75 mL, 1.0 mL and 1.5 mL of 1,2-dichloroethane provided 4a with an enhanced enantioselectivity of 92% ee, 93% ee and 95% ee, respectively (Table 2, entries 3–5). On performing the reaction in 2.0 mL of 1,2-dichloroethane, 4a was obtained in 88% yield and increased enantioselectivity of 96% ee after a long reaction time of 96 hours (Table 2, entry 6). There was a small difference in the enantioselectivity of the product 4a on performing the reaction in 1.5 mL and 2.0 mL of 1,2-dichloroethane, but the rate of the reaction was faster when a lower amount of solvent was used. So, we decided to employ 1.5 mL of 1,2-dichloroethane as a solvent for the further optimization experiments.
Table 2: Effect of the amount of solvent (DCE) on the enantioselectivity of the Michael addition of acetone (2a) to isatylidenemalononitrile (3a) catalyzed by 1a TCA.a
| Entry | Amount of solvent (mL) | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 0.25 | 24 | 93 | 91 |
| 2 | 0.50 | 26 | 93 | 91 |
| 3 | 0.75 | 32 | 92 | 92 |
| 4 | 1.00 | 40 | 90 | 93 |
| 5 | 1.50 | 60 | 90 | 95 |
| 6 | 2.00 | 96 | 88 | 96 |
aReaction conditions: 1.5 mmol of acetone (2a), 0.125 mmol of isatylidenemalononitrile (3a), 1,2-dichloroethane (0.25–2.00 mL), 10 mol % of catalyst 1a, 10 mol % of TCA, at 25 °C. bIsolated yield determined after chromatographic purification. cEnantiomeric excess determined by chiral HPLC.
In order to obtain a highly enantioselective transformation, the effect of different acid additives on the enantioselectivity of 4a was studied (Table 3). The reaction was performed with 3,5-dinitrobenzoic acid and chloroacetic acid afforded 4a in good yield of 91% and 90%; and moderate enantioselectivity of 58% ee and 60% ee, respectively (Table 3, entries 1 and 2). The reaction carried out with strong acids such as trifluoromethanesulfonic acid (TsOH) and trifluoroacetic acid (TFA) gave 4a in good yields of 86% and 89% and high enantioselectivities of 97% ee and 96% ee (Table 3, entries 3 and 4). The application of L-camphorsulfonic acid as an additive resulted in the isolation of 4a in 92% yield and an enantioselectivity of 98% ee after 30 hours (Table 3, entry 5). The D-camphorsulfonic acid turned out to be the best acid additive providing 4a in high yield of 93% and excellent enantioselectivity of 99% ee after a reaction time of 24 hours (Table 3, entry 6) [46].
Table 3: Additive screening of the 1a catalyzed Michael addition of acetone (2a) to isatylidenemalononitrile (3a)a.
| Entry | Additive | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 3,5-dinitrobenzoic acid | 36 | 91 | 58 |
| 2 | chloroacetic acid | 36 | 90 | 60 |
| 3 | TsOH | 60 | 86 | 97 |
| 4 | TFA | 48 | 89 | 96 |
| 5 | L-CSA | 30 | 92 | 98 |
| 6 | D-CSA | 24 | 93 | 99 |
aReaction conditions: 1.5 mmol of acetone (2a), 0.125 mmol of isatylidenemalononitrile (3a), 1.5 mL of DCE, 10 mol % of catalyst 1a, 10 mol % of additive at 25 °C. bIsolated yield determined after chromatographic purification. cEnantiomeric excess determined by chiral HPLC.
Even though an excellent level of enantioselectivity of the product was observed with catalyst 1a we screened different diamine catalysts in our quest for a superior catalyst. L-Isoleucine derived primary-tertiary diamine catalysts having piperidinyl 1b and pyrrolidinyl 1c groups gave Michael product 4a in high yield (>90%) and excellent enantioselectivity (98% ee each) (Table 4, entries 2 and 3). The primary-tertiary diamine catalysts characterized by an acyclic tertiary amine, such as a N,N-dioctyl (1d) group, gave 4a in 89% yield and 96% ee (Table 4, entry 4). The L-leucine, L-valine and L-phenylalanine derived primary-tertiary diamine catalysts (1e–1g) also provide the Michael adduct 4a in good yield (92–94%) and an excellent level of enantioselectivity (97–98% ee) (Table 4, entries 5–7). All primary-tertiary diamine catalysts 1a–1g gave 4a in high yield (89–94%) with an excellent level of enantioselectivity (96–99% ee). In contrast, the primary-secondary diamine 1h catalyst afforded Michael adduct 4a in 10% yield after a long reaction time (Table 4, entry 8). The screening study highlights the importance of the primary-tertiary diamine skeleton in the catalysis of the addition of acetone (2a) to isatylidenemalononitrile (3a). Thus, the best reaction conditions consist of 10 mol % of catalyst 1a, 10 mol % of D-camphorsulfonic acid as an additive and 1.5 mL of 1,2-dichloroethane at room temperature providing Michael adduct 4a in 93% yield and 99% ee.
Table 4: Catalyst screening of the enantioselective Michael addition of acetone (2a) to isatylidenemalononitrile (3a).a
| Entry | Catalyst | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 1a | 24 | 93 | 99 |
| 2 | 1b | 24 | 93 | 98 |
| 3 | 1c | 24 | 91 | 98 |
| 4 | 1d | 30 | 89 | 96 |
| 5 | 1e | 30 | 93 | 98 |
| 6 | 1f | 24 | 94 | 98 |
| 7 | 1g | 30 | 92 | 97 |
| 8 | 1h | 96 | 10 | – |
aReaction conditions: 1.5 mmol of acetone (2a), 0.125 mmol of isatylidenemalononitrile (3a), 1.5 mL of 1,2-dichloroethane, 10 mol % of catalyst 1a–1h, 10 mol % of D-CSA at 25 °C. bIsolated yield determined after chromatographic purification. cEnantiomeric excess determined by chiral HPLC.
Once the optimized reaction conditions have been found, the substrate scope was investigated by using different ketones 2a–c and isatylidenemalononitrile derivatives 3a–i (Scheme 2). The methodology was found to be suitable for both N-substituted isatylidenemalononitrile and N–H isatylidenemalononitrile derivatives. Acetone (2a) reacts well with various N–H isatylidenemalononitrile derivatives (3a–f) providing corresponding Michael adducts 4a–f in excellent enantioselectivity (96–99% ee) after a reaction time of 24–36 hours. The 5-fluoroisatylidenemalononitrile (3b), 5-chloroisatylidenemalononitrile (3c), 5-bromoisatylidenemalononitrile (3d) and 5-iodoisatylidenemalononitrile (3e) gave corresponding Michael adducts 4b–e in 93%, 95%, 94% and 91% yield and 98% ee, 98% ee, 98% ee and 99% ee, respectively (Table 5, entries 2–5). The reaction of 5,7-dibromoisatylidenemalononitrile (3f) with acetone (2a) proceeds with a high yield of 92% and a high enantioselectivity of 96% ee (Table 5, entry 6). The N-substituted isatylidenemalononitriles 3g–i react slowly with acetone (2a) to afford Michael adducts 4g–i in good yield (85–87%) and good enantioselectivity (88–92% ee) (Table 5, entries 7–9). Acetone (2a) reacts well with N-allyl isatylidenemalononitrile derivatives 3g and 3h to provide the respective Michael adducts 4g and 4h in 85% and 87% yield and 89% ee and 92% ee, respectively (Table 5, entries 7 and 8). Using N-benzyl isatylidenemalononitrile (3i), the Michael adduct 4i was isolated in 86% yield and 88% ee (Table 5, entry 9). A recently reported similar reaction catalyzed by Cinchona alkaloid-based primary amine catalyst requires high catalyst loading and is only suitable for N-unprotected isatylidenemalononitrile derivatives [5]. In contrast, our methodology is suitable for both N-unprotected and N-protected isatylidenemalononitrile derivatives and is highly enantioselective. Next, the substrate scope of the reaction was extended to different acyclic ketones 2b and 2c. Under the optimized conditions, the Michael addition of methyl isobutyl ketone (2b) and 2-octanone (2c) with 3a provided Michael adducts 4j and 4k in 24% and 41% yield and 97% ee and 96% ee, respectively (Table 5, entries 10 and 11). Due to the low reactivity of these ketones, these experiments were carried out after a slight modification of the optimized conditions, i.e., a higher catalyst loading of 20 mol % of 1a and 1.0 mL of 1,2-dichloroethane as a solvent. The 20 mol % of 1a catalyzes the Michael addition of 2b with 3a providing Michael adduct 4j in 80% yield and 96% ee after a reaction time of 7 days (Table 5, entry 12). The 2-octanone (2c) gave Michael adduct 4k in 85% yield and 97% ee after a reaction time of 6 days (Table 5, entry 13). The R absolute configuration of Michael adducts was assigned by comparing the HPLC chromatograms of Michael adducts with that reported in the literature [23,24].
![[1860-5397-10-91-i2]](/bjoc/content/inline/1860-5397-10-91-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the addition of 2 with 3 catalyzed by 1a D-CSA.
Scheme 2: Substrate scope of the addition of 2 with 3 catalyzed by 1a D-CSA.
Table 5: Substrate scope of 1a D-CSA catalyzed asymmetric Michael reaction of ketones 2 with isatylidenemalononitrile derivatives 3.a
| Entry | 2 | 3 | Time (h) | 4 | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 2a | 3a | 26 | 4a | 92 | 99 |
| 2 | 2a | 3b | 26 | 4b | 93 | 98 |
| 3 | 2a | 3c | 26 | 4c | 95 | 98 |
| 4 | 2a | 3d | 30 | 4d | 94 | 98 |
| 5 | 2a | 3e | 36 | 4e | 91 | 99 |
| 6 | 2a | 3f | 24 | 4f | 92 | 96 |
| 7 | 2a | 3g | 72 | 4g | 85 | 89 |
| 8 | 2a | 3h | 78 | 4h | 87 | 92 |
| 9 | 2a | 3i | 78 | 4i | 86 | 88 |
| 10 | 2b | 3a | 168 | 4j | 24 | 97 |
| 11 | 2c | 3a | 168 | 4k | 41 | 96 |
| 12d | 2b | 3a | 168 | 4j | 80 | 96 |
| 13d | 2c | 3a | 144 | 4k | 85 | 97 |
aReaction conditions: 1.5 mmol of ketones 2, 0.125 mmol of isatylidenemalononitrile derivatives 3, 1.5 mL of 1,2-dichloroethane, 10 mol % of catalyst 1a, 10 mol % of additive D-CSA at 25 °C. bIsolated yield determined after chromatographic purification. cEnantiomeric excess determined by chiral HPLC. dThe reaction is performed with 20 mol % catalyst 1a and 1.0 mL of 1,2-dichloroethane.
Next, we studied the multicomponent version of this reaction. Acetone, isatin and malononitrile react in one pot providing Michael product 4a in a good yield of 80% and a high enantioselectivity of 98% ee (Scheme 3). The slightly lower yield of the one-pot process compared to the stepwise process was due to the competing reaction of isatin and acetone to form aldol adduct 5 (8% yield). The reaction involves the initial formation of isatylidenemalononitrile by Knoevenagel condensation of isatin with malononitrile followed by the addition of acetone to provide Michael adduct 4a. Thus, catalyst 1a also finds its successful application in the multicomponet version of this reaction without compromising enantioselectivity – albeit with a slight loss in yield.
![[1860-5397-10-91-i3]](/bjoc/content/inline/1860-5397-10-91-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: One-pot, three-component Knoevenagel condensation–Michael addition.
Scheme 3: One-pot, three-component Knoevenagel condensation–Michael addition.
We further demonstrated that Michael adducts could be transformed into spirooxindoles by following a simple strategy. The reduction of Michael adduct 4a with sodium borohydride in ethanol followed by spontaneous cyclization gave spirooxindole product 6 in 90% yield, 82:18 dr and 98% ee (Scheme 4).
![[1860-5397-10-91-i4]](/bjoc/content/inline/1860-5397-10-91-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cascade reduction–cyclization for the synthesis of spirooxindole.
Scheme 4: Cascade reduction–cyclization for the synthesis of spirooxindole.
Conclusion
In conclusion, we successfully demonstrated the use of the very simple primary-tertiary diamine catalyst 1a in combination with D-CSA as an additive for the enantioselective catalysis of a Michael addition of acetone to isatylidenemalononitriles. A three component process involving a domino Knoevenagel–Michael sequence was developed. 3,3'-Disubstituted oxindole could be transformed into spirooxindoles by reduction with NaBH4.
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of 1H and 13C NMR spectra of Michael adducts, and HPLC chromatogram of products 4. | ||
| Format: PDF | Size: 1.1 MB | Download |
Acknowledgements
We are grateful for the financial support from CSIR, India to SSC (research Grant No. 02(0009)/11/EMR-II) and a RA fellowship to AK. Financial support from the Department of Science and Technology (DST), India under the FIST program and UGC, India, under CAS-I is gratefully acknowledged.
References
-
Jha, S. C.; Joshi, N. N. ARKIVOC 2002, No. vii, 167–196. doi:10.3998/ark.5550190.0003.718
Return to citation in text: [1] [2] -
Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877–1894. doi:10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U
Return to citation in text: [1] [2] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] [2] -
Almasi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] [2] -
Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747
Return to citation in text: [1] [2] [3] -
Bhanja, C.; Jena, S.; Nayak, S.; Mohapatra, S. Beilstein J. Org. Chem. 2012, 8, 1668–1694. doi:10.3762/bjoc.8.191
Return to citation in text: [1] [2] -
Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h
Return to citation in text: [1] [2] -
Chauhan, P.; Kaur, J.; Chimni, S. S. Chem.–Asian J. 2013, 8, 328–346. doi:10.1002/asia.201200684
Return to citation in text: [1] [2] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] [2] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161
Return to citation in text: [1] -
Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821–6841. doi:10.1002/ejoc.201100836
Return to citation in text: [1] -
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e
Return to citation in text: [1] -
Shen, K.; Liu, X.; Lin, L.; Feng, X. Chem. Sci. 2012, 3, 327–334. doi:10.1039/c1sc00544h
Return to citation in text: [1] -
Arai, T.; Yamamoto, Y.; Awata, A.; Kamiya, K.; Ishibashi, M.; Arai, M. A. Angew. Chem., Int. Ed. 2013, 52, 2486–2490. doi:10.1002/anie.201208918
Return to citation in text: [1] -
Peddibhotla, S. Curr. Bioact. Compd. 2009, 5, 20–38. doi:10.2174/157340709787580900
Return to citation in text: [1] -
Chen, W.-B.; Wu, Z.-J.; Pei, Q.-L.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2010, 12, 3132–3135. doi:10.1021/ol1009224
Return to citation in text: [1] -
Lan, Y.-B.; Zhao, H.; Liu, Z.-M.; Liu, G.-G.; Tao, J.-C.; Wang, X.-W. Org. Lett. 2011, 13, 4866–4869. doi:10.1021/ol201943g
Return to citation in text: [1] -
Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094
Return to citation in text: [1] -
Liu, L.; Wu, D.; Zheng, S.; Li, T.; Li, X.; Wang, S.; Li, J.; Li, H.; Wang, W. Org. Lett. 2012, 14, 134–137. doi:10.1021/ol202931e
Return to citation in text: [1] -
Wei, W.-T.; Chen, C.-X.; Lu, R.-J.; Wang, J.-J.; Zhang, X.-J.; Yan, M. Org. Biomol. Chem. 2012, 10, 5245–5252. doi:10.1039/c2ob25629k
Return to citation in text: [1] -
Liu, L.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Commun. 2012, 48, 1692–1694. doi:10.1039/c2cc17067a
Return to citation in text: [1] [2] -
Zhao, H.; Lan, Y.-B.; Liu, Z.-M.; Wang, Y.; Wang, X.-W.; Tao, J.-C. Eur. J. Org. Chem. 2012, 1935–1944. doi:10.1002/ejoc.201101810
Return to citation in text: [1] [2] -
Xu, L.-W.; Lu, Y. Org. Biomol. Chem. 2008, 6, 2047–2053. doi:10.1039/b803116a
Return to citation in text: [1] -
Peng, F.; Shao, Z. J. Mol. Catal. A: Chem. 2008, 285, 1–13. doi:10.1016/j.molcata.2007.12.027
Return to citation in text: [1] -
Chen, Y.-C. Synlett 2008, 1919–1930. doi:10.1055/s-2008-1078524
Return to citation in text: [1] -
Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807–1821. doi:10.1039/b821070e
Return to citation in text: [1] -
Jiang, L.; Chen, Y.-C. Catal. Sci. Technol. 2011, 1, 354–365. doi:10.1039/c0cy00096e
Return to citation in text: [1] -
Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036
Return to citation in text: [1] -
Xu, Y.; Córdova, A. Chem. Commun. 2006, 460–462. doi:10.1039/B514783M
Return to citation in text: [1] -
Yang, Y.-Q.; Zhao, G. Chem.–Eur. J. 2008, 14, 10888–10891. doi:10.1002/chem.200801749
Return to citation in text: [1] -
Mao, Z.; Jia, Y.; Li, W.; Wang, R. J. Org. Chem. 2010, 75, 7428–7430. doi:10.1021/jo101188m
Return to citation in text: [1] -
Yang, Y.-Q.; Chen, X.-K.; Xiao, H.; Liu, W.; Zhao, G. Chem. Commun. 2010, 46, 4130–4132. doi:10.1039/c002552f
Return to citation in text: [1] -
Lalonde, M. P.; Chen, Y.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 6366–6370. doi:10.1002/anie.200602221
Return to citation in text: [1] -
Kokotos, C. G.; Kokotos, G. Adv. Synth. Catal. 2009, 351, 1355–1362. doi:10.1002/adsc.200800812
Return to citation in text: [1] -
Serdyuk, O. V.; Heckel, C. M.; Tsogoeva, S. B. Org. Biomol. Chem. 2013, 11, 7051–7071. doi:10.1039/c3ob41403e
Return to citation in text: [1] -
Yu, F.; Hu, H.; Gu, X.; Ye, J. Org. Lett. 2012, 14, 2038–2041. doi:10.1021/ol300489q
See for Michael reaction via iminium activation.
Return to citation in text: [1] -
Luo, S.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.-P. J. Am. Chem. Soc. 2007, 129, 3074–3075. doi:10.1021/ja069372j
Return to citation in text: [1] -
Raj, M.; Parashari, G. S.; Singh, V. K. Adv. Synth. Catal. 2009, 351, 1284–1288. doi:10.1002/adsc.200900122
Return to citation in text: [1] -
Li, J.; Fu, N.; Li, X.; Luo, S.; Cheng, J.-P. J. Org. Chem. 2010, 75, 4501–4507. doi:10.1021/jo100976e
Return to citation in text: [1] -
Demuynck, A. L. W.; Vanderleyden, J.; Sels, B. F. Adv. Synth. Catal. 2010, 352, 2421–2426. doi:10.1002/adsc.201000419
Return to citation in text: [1] -
Jiang, Z.; Lu, Y. Tetrahedron Lett. 2010, 51, 1884–1886. doi:10.1016/j.tetlet.2010.02.044
Return to citation in text: [1] -
Kumar, A.; Singh, S.; Kumar, V.; Chimni, S. S. Org. Biomol. Chem. 2011, 9, 2731–2742. doi:10.1039/c0ob00898b
Return to citation in text: [1] -
Kumar, A.; Chimni, S. S. Tetrahedron 2013, 69, 5197–5204. doi:10.1016/j.tet.2013.04.044
Return to citation in text: [1] -
Liu, C.; Zhu, Q.; Huang, K.-W.; Lu, Y. Org. Lett. 2011, 13, 2638–2641. doi:10.1021/ol200747x
See for primary amine/CSA ion pair in enamine catalysis.
Return to citation in text: [1]
| 1. | Jha, S. C.; Joshi, N. N. ARKIVOC 2002, No. vii, 167–196. doi:10.3998/ark.5550190.0003.718 |
| 2. | Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877–1894. doi:10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U |
| 3. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 4. | Almasi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 5. | Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747 |
| 6. | Bhanja, C.; Jena, S.; Nayak, S.; Mohapatra, S. Beilstein J. Org. Chem. 2012, 8, 1668–1694. doi:10.3762/bjoc.8.191 |
| 7. | Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h |
| 8. | Chauhan, P.; Kaur, J.; Chimni, S. S. Chem.–Asian J. 2013, 8, 328–346. doi:10.1002/asia.201200684 |
| 23. | Liu, L.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Commun. 2012, 48, 1692–1694. doi:10.1039/c2cc17067a |
| 24. | Zhao, H.; Lan, Y.-B.; Liu, Z.-M.; Wang, Y.; Wang, X.-W.; Tao, J.-C. Eur. J. Org. Chem. 2012, 1935–1944. doi:10.1002/ejoc.201101810 |
| 23. | Liu, L.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Commun. 2012, 48, 1692–1694. doi:10.1039/c2cc17067a |
| 24. | Zhao, H.; Lan, Y.-B.; Liu, Z.-M.; Wang, Y.; Wang, X.-W.; Tao, J.-C. Eur. J. Org. Chem. 2012, 1935–1944. doi:10.1002/ejoc.201101810 |
| 18. | Chen, W.-B.; Wu, Z.-J.; Pei, Q.-L.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2010, 12, 3132–3135. doi:10.1021/ol1009224 |
| 19. | Lan, Y.-B.; Zhao, H.; Liu, Z.-M.; Liu, G.-G.; Tao, J.-C.; Wang, X.-W. Org. Lett. 2011, 13, 4866–4869. doi:10.1021/ol201943g |
| 20. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 21. | Liu, L.; Wu, D.; Zheng, S.; Li, T.; Li, X.; Wang, S.; Li, J.; Li, H.; Wang, W. Org. Lett. 2012, 14, 134–137. doi:10.1021/ol202931e |
| 22. | Wei, W.-T.; Chen, C.-X.; Lu, R.-J.; Wang, J.-J.; Zhang, X.-J.; Yan, M. Org. Biomol. Chem. 2012, 10, 5245–5252. doi:10.1039/c2ob25629k |
| 17. | Peddibhotla, S. Curr. Bioact. Compd. 2009, 5, 20–38. doi:10.2174/157340709787580900 |
| 46. |
Liu, C.; Zhu, Q.; Huang, K.-W.; Lu, Y. Org. Lett. 2011, 13, 2638–2641. doi:10.1021/ol200747x
See for primary amine/CSA ion pair in enamine catalysis. |
| 9. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050 |
| 10. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 11. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975 |
| 12. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161 |
| 13. | Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821–6841. doi:10.1002/ejoc.201100836 |
| 14. | Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e |
| 15. | Shen, K.; Liu, X.; Lin, L.; Feng, X. Chem. Sci. 2012, 3, 327–334. doi:10.1039/c1sc00544h |
| 16. | Arai, T.; Yamamoto, Y.; Awata, A.; Kamiya, K.; Ishibashi, M.; Arai, M. A. Angew. Chem., Int. Ed. 2013, 52, 2486–2490. doi:10.1002/anie.201208918 |
| 5. | Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747 |
| 39. | Luo, S.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.-P. J. Am. Chem. Soc. 2007, 129, 3074–3075. doi:10.1021/ja069372j |
| 40. | Raj, M.; Parashari, G. S.; Singh, V. K. Adv. Synth. Catal. 2009, 351, 1284–1288. doi:10.1002/adsc.200900122 |
| 41. | Li, J.; Fu, N.; Li, X.; Luo, S.; Cheng, J.-P. J. Org. Chem. 2010, 75, 4501–4507. doi:10.1021/jo100976e |
| 42. | Demuynck, A. L. W.; Vanderleyden, J.; Sels, B. F. Adv. Synth. Catal. 2010, 352, 2421–2426. doi:10.1002/adsc.201000419 |
| 43. | Jiang, Z.; Lu, Y. Tetrahedron Lett. 2010, 51, 1884–1886. doi:10.1016/j.tetlet.2010.02.044 |
| 44. | Kumar, A.; Singh, S.; Kumar, V.; Chimni, S. S. Org. Biomol. Chem. 2011, 9, 2731–2742. doi:10.1039/c0ob00898b |
| 45. | Kumar, A.; Chimni, S. S. Tetrahedron 2013, 69, 5197–5204. doi:10.1016/j.tet.2013.04.044 |
| 31. | Xu, Y.; Córdova, A. Chem. Commun. 2006, 460–462. doi:10.1039/B514783M |
| 32. | Yang, Y.-Q.; Zhao, G. Chem.–Eur. J. 2008, 14, 10888–10891. doi:10.1002/chem.200801749 |
| 33. | Mao, Z.; Jia, Y.; Li, W.; Wang, R. J. Org. Chem. 2010, 75, 7428–7430. doi:10.1021/jo101188m |
| 34. | Yang, Y.-Q.; Chen, X.-K.; Xiao, H.; Liu, W.; Zhao, G. Chem. Commun. 2010, 46, 4130–4132. doi:10.1039/c002552f |
| 35. | Lalonde, M. P.; Chen, Y.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 6366–6370. doi:10.1002/anie.200602221 |
| 36. | Kokotos, C. G.; Kokotos, G. Adv. Synth. Catal. 2009, 351, 1355–1362. doi:10.1002/adsc.200800812 |
| 37. | Serdyuk, O. V.; Heckel, C. M.; Tsogoeva, S. B. Org. Biomol. Chem. 2013, 11, 7051–7071. doi:10.1039/c3ob41403e |
| 10. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 25. | Xu, L.-W.; Lu, Y. Org. Biomol. Chem. 2008, 6, 2047–2053. doi:10.1039/b803116a |
| 26. | Peng, F.; Shao, Z. J. Mol. Catal. A: Chem. 2008, 285, 1–13. doi:10.1016/j.molcata.2007.12.027 |
| 27. | Chen, Y.-C. Synlett 2008, 1919–1930. doi:10.1055/s-2008-1078524 |
| 28. | Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807–1821. doi:10.1039/b821070e |
| 29. | Jiang, L.; Chen, Y.-C. Catal. Sci. Technol. 2011, 1, 354–365. doi:10.1039/c0cy00096e |
| 30. | Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036 |
| 1. | Jha, S. C.; Joshi, N. N. ARKIVOC 2002, No. vii, 167–196. doi:10.3998/ark.5550190.0003.718 |
| 2. | Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877–1894. doi:10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U |
| 3. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 4. | Almasi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 5. | Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747 |
| 6. | Bhanja, C.; Jena, S.; Nayak, S.; Mohapatra, S. Beilstein J. Org. Chem. 2012, 8, 1668–1694. doi:10.3762/bjoc.8.191 |
| 7. | Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h |
| 8. | Chauhan, P.; Kaur, J.; Chimni, S. S. Chem.–Asian J. 2013, 8, 328–346. doi:10.1002/asia.201200684 |
| 38. |
Yu, F.; Hu, H.; Gu, X.; Ye, J. Org. Lett. 2012, 14, 2038–2041. doi:10.1021/ol300489q
See for Michael reaction via iminium activation. |
© 2014 Kumar and Chimni; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)