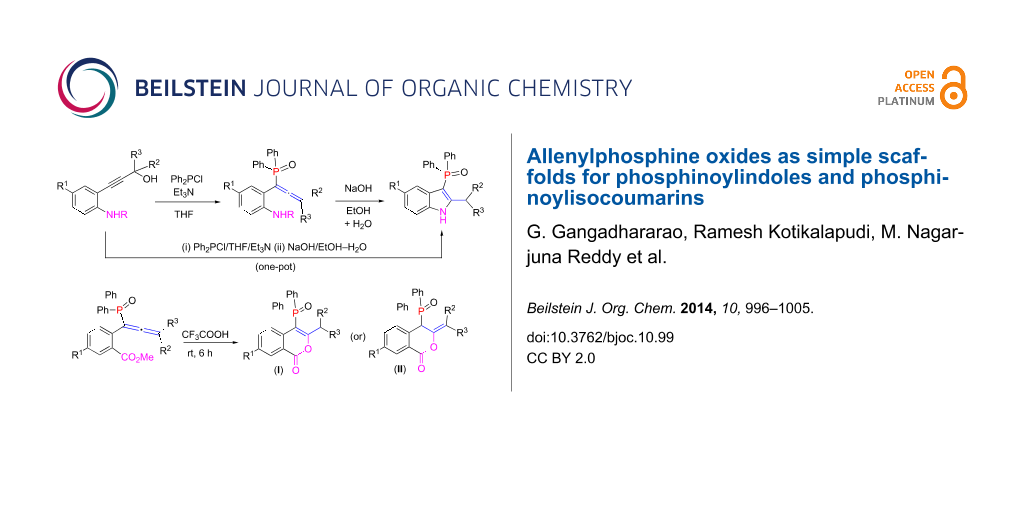
Abstract
A range of phosphinoylindoles was prepared in one-pot from functionalized propargyl alcohols and a suitable P(III) precursor via a base-mediated reaction. The reaction proceeds via the intermediacy of allenylphosphine oxides. Similarly, phosphinoylisocoumarins were prepared from allenylphosphine oxides in a trifluoroacetic acid-mediated reaction; the latter also acts as a solvent. Interestingly, in the presence of wet trifluoroacetic acid, in addition to phosphinoylisocoumarins, phosphorus-free isocoumarins were also obtained. Key products were characterized by single crystal X-ray crystallography.

Graphical Abstract
Introduction
Allenes, by virtue of cumulative double bonds that facilitate reactions with diverse classes of substrates, are versatile building blocks from a synthetic perspective [1,2]. They are also found in many natural products, pharmaceuticals [3] and molecular materials [4]. Thus, over the last decade, allenes have attained a prominent position in organic transformations like cycloaddition, cycloisomerization, base or metal-catalyzed reactions [5-7]. In particular, cyclization reaction of allenes has emerged as a valuable tool in developing different methods leading to various carbo-/heterocycles [8-14]. Allenylphosphonates and allenylphosphine oxides, as a subclass of allenes, have also been utlized in several novel transformations [15-17]. It may also be noted that organophosphonates in addition have wide applications in medicinal chemistry [3,18,19] and as reagents in organic synthesis [20]. In our previous reports, we described the utility of phosphorus-based allenes in various cyclization reactions involving heteroatoms that could lead to phosphono/phosphinoyl hetero-/carbocycles [21-30]. The reported series include phosphonobenzofurans/indenones [21,22], -pyrazoles [23], -chromenes/thiochromenes [24,25], -pyrroles [26], multiply substituted furans [27], indolopyran-1-ones [28], N-hydroxyindolinones [29], and oxindoles [30]. In the reaction shown in Scheme 1a, for the formation of the phosphinoylindolinone, one of the oxygen atoms of the nitro group has been moved to a carbon [29]. The reaction shown in Scheme 1b led to rather previously unsuspected and unexpected benzazepines as products. After the elimination of a CO2 molecule, this reaction also features an unprecedented rearrangement involving the interemdiate allene [29]. Many other unusual transformations have also been reported recently [31]. In another reaction leading to phosphinoylindenone depicted in Scheme 1c, an intramolecular ene-reaction is possibly involved and in Scheme 1d the reaction led to phosphinoylisochromenes via deprotection of an allene intermediate under Lewis acid mediation [22]. In this context it was of interest to see, in a reaction like that shown in Scheme 1c, whether the introduction of an amide or a carboxylate ester in place of the –CHO group could lead to phosphinoyl-subtstituted indoles/isocoumarins via allenic intermediates or not. It is pertinent to note that indoles and isocoumarins are core structures found in many natural and pharmacological products [32-34]. Thus in this paper, we wish to report simple synthetic routes to phosphinoylindoles, and -isocoumarins utilizing functionalized allenylphosphine oxides/allenylphosphonates.
![[1860-5397-10-99-i1]](/bjoc/content/inline/1860-5397-10-99-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of P(III)-Cl precursors with propargyl alcohols leading to phosphorus based (a) N-hydroxyindolinone, (b) benzazepine, (c) indenone and (d) isochromene via allenic intermediates.
Scheme 1: Reaction of P(III)-Cl precursors with propargyl alcohols leading to phosphorus based (a) N-hydroxyi...
Results and Discussion
In order to achieve the anticipated phosphinoylindoles/isocoumarins, we prepared a variety of functionalized propargyl alcohols 1a–m and 2a–j containing an acetamide, benzamide or an ester group at the ortho position (Figure 1) [35-37]. Some of the propargyl alcohols 1a–c, 1m and 2a–j were transformed to allenylphosphine oxides 3a–c, 3m and 4a–j (Scheme 2) by following known methods [38,39].
![[1860-5397-10-99-1]](/bjoc/content/figures/1860-5397-10-99-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Functionalized propargyl alcohols 1a–m and 2a–j used in the present study.
Figure 1: Functionalized propargyl alcohols 1a–m and 2a–j used in the present study.
![[1860-5397-10-99-i2]](/bjoc/content/inline/1860-5397-10-99-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of functionalized allenes 3a–c, 3m and 4a–j.
Scheme 2: Synthesis of functionalized allenes 3a–c, 3m and 4a–j.
After having several functionalized allenes in hand, initially we chose allenes 3a and 3m to achieve intramolecular cyclization. These were treated with 0.5 mol equivalents of base (K3PO4) since the substrates contain active hydrogen. This reaction afforded the N-substituted phosphinoylindoles 5 and 7, 8. Essentially a single isomer 5 (a dihydroindole), in which the N–H proton moves only to the α-carbon resulting in an exocyclic double bond, was formed (Scheme 3). The presence of a doublet for PCH carbon at δ 48.3 with a 1J(P–C) value of 62.0 Hz reveals that the phosphorus moiety is attached to an sp3-hybridized carbon. On the other hand, in the reaction using the =CHMe allene 3m, two isomers in which the N–H proton moves to either the α-carbon (7) or the γ-carbon (8), are obtained. These two isomers can be readily distinguished by the corresponding δ and 1J values for the P–C carbon (for 7, δ 47.3 and J = 62.0 Hz; for 8, δ 106.5 and J = 120.0 Hz). Overall, the yields of the isolated products were excellent in both cases. The structure of compound 7 was further confirmed by X-ray crystallography (Figure 2). The C=CHMe distance of 1.317(2) Å clearly indicates a double bond between these two carbon atoms. The other stereoisomer in which the methyl group is trans to the nitrogen was not observed. Interestingly though, the removal of the acyl/benzoyl group on the nitrogen in compounds 5 or 7, 8 in aq NaOH afforded the 2,3-disubstituted NH-indoles 6 or 9, respectively, in excellent yields. The NH band (3156 cm−1) in the IR spectrum and a doublet for PC carbon at δ 98.4 (1J(PC) = 128.0 Hz) reveal the identity of compound 9. Its structure was further confirmed by X-ray crystallography (Figure 3).
![[1860-5397-10-99-i3]](/bjoc/content/inline/1860-5397-10-99-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of functionalized allenes 3a and 3m leading to phosphinoylindoles. Conditions: (i) K3PO4 (0.5 equiv), THF, 80 °C, 12 h, (ii) NaOH (2 equiv), EtOH/H2O (4:1), 80 °C, 8 h.
Scheme 3: Reaction of functionalized allenes 3a and 3m leading to phosphinoylindoles. Conditions: (i) K3PO4 (...
![[1860-5397-10-99-2]](/bjoc/content/figures/1860-5397-10-99-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of compound 7. Hydrogen atoms (except PCH) are omitted for clarity. Selected bond distances (Å): P1–C13 1.8402(15), C13–C20 1.5161(19), C13–C14 1.512(2), C20–N1 1.4508(18), C20–C21 1.317(2).
Figure 2: Molecular structure of compound 7. Hydrogen atoms (except PCH) are omitted for clarity. Selected bo...
![[1860-5397-10-99-3]](/bjoc/content/figures/1860-5397-10-99-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of compound 9. Hydrogen atoms (except NH) are omitted for clarity. Selected bond distances (Å): P1–C13 1.771(2), C13–C20 1.385(3), C13–C14 1.450(3), C20–N1 1.360(3), C20–C21 1.489(3).
Figure 3: Molecular structure of compound 9. Hydrogen atoms (except NH) are omitted for clarity. Selected bon...
Subsequently, we used aq sodium hydroxide as the base instead of K3PO4 (cf. conditions (ii) in Scheme 3) to perform the reaction on allene 3a. To our delight, only phosphinoyl-NH-indole 6 was the sole product with not even traces of 5 (Scheme 4). This shows that a strong base like sodium hydroxide effectively performs both deprotection and cyclization in a single step.
![[1860-5397-10-99-i4]](/bjoc/content/inline/1860-5397-10-99-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of phosphinoylindole from allene 3a in a single step.
Scheme 4: Synthesis of phosphinoylindole from allene 3a in a single step.
With the above conditions in hand, we then performed the reaction in one pot starting from propargyl alcohol 1a without isolating the intermediate allenylphosphine oxide 3a. Gratifyingly, the method furnished the desired product 6 in 80% yield. Inspired by this, functionalized propargyl alcohols 1b–l were also subjected to the same one-pot conditions (Scheme 5). This one-pot strategy furnished the desired phosphinoylindoles 9–19 in good to excellent yields without any difficulty in isolation. Analogous products could also be isolated using the P(III) precursor (OCH2CMe2CH2O)PCl (see Supporting Information File 1 for details). In our attmept to obtain phosphorus-free 2-alkylindole from 17 in the presence of triflic acid (as a solvent; 100 °C) led to a mixture of products in which the benzyl group also was cleaved (NMR evidence). Such a reductive cleavage of the P–C bond from phosphinoyl indoles is a reaction that we are still exploring.
![[1860-5397-10-99-i5]](/bjoc/content/inline/1860-5397-10-99-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: One-pot preparation of substituted phosphinoylindoles 6 and 9–19 from functionalized alcohols.
Scheme 5: One-pot preparation of substituted phosphinoylindoles 6 and 9–19 from functionalized alcohols.
A plausible pathway for the formation of phosphinoylindoles 6 and 9–19 is shown in Scheme 6. As depicted above in Scheme 2, the normal reaction of propargyl alcohol with chlorodiphenylphosphine is expected to lead to the allenylphosphine oxide. We believe that there is a subtle difference between the use of K3PO4 and aq NaOH. K3PO4 abstracts the NH proton from allenylphosphine oxide leading to intermediate I which is followed by attack of the nitrogen lone pair on the β-carbon [24] of the allene forming addition product II or III. This upon treating with aq NaOH leads to the deacylated/debenzoylated phosphinoylindoles. In the one-pot reaction, though, the in situ generated allenylphosphine oxide first undergoes deacylation/debenzoylation with aq NaOH resulting in –NH2 functionalized allene IV; the lone pair on nitrogen will then attack the β-carbon of the allene intramolecularly leading to phosphinoylindoles 6 or 9–19.
![[1860-5397-10-99-i6]](/bjoc/content/inline/1860-5397-10-99-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible pathway for the formation of phosphinoyl indoles 6 and 9–19.
Scheme 6: Possible pathway for the formation of phosphinoyl indoles 6 and 9–19.
After succeeding in generating phosphinoylindoles, we then concentrated on synthesizing phosphinoylisocoumarins. To achieve this, we treated the functionalized allene precursors 4a–j that are tethered with a methyl ester group, with an excess of trifluoroacetic acid at room temperature for 6 h. Gratifyingly, this readily leds to the phosphinoylisocoumarins 20–29 (Scheme 7) in good yields. In the case of compound 25, as expected, both the E and Z isomers are present in a ratio of 1:0.65 (close Rf values). Very subtle energy differences seem to be prevalent between the dihydroisocoumains 22, 24, 25, 28, 29 and the normal isocoumarins 20, 21, 23, 26, 27. The former set shows a doublet in the 1H NMR spectra at δ ~ 4.78 (2J(P–H) = 18.0 Hz, PCH) which is absent in the latter set; the difference in the value of 1J(P–C) in the two sets is also consistent with the hybridization at the corresponding α-carbon (to phosphorus). Finally, the X-ray structure was determined for 20 (Figure 4).
![[1860-5397-10-99-i7]](/bjoc/content/inline/1860-5397-10-99-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of phosphinoylisocoumarins from functionalized allenes.
Scheme 7: Synthesis of phosphinoylisocoumarins from functionalized allenes.
![[1860-5397-10-99-4]](/bjoc/content/figures/1860-5397-10-99-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure of 20. Selected bond lengths [Å] with estimated standard deviations are given in parentheses: O3–C21 1.386(2), C21–C22 1.486(3).
Figure 4: Molecular structure of 20. Selected bond lengths [Å] with estimated standard deviations are given i...
The above reaction is believed to proceed by the initial interaction of H+ with the α,β-allenic double bond to lead to V (Scheme 8) which on subsequent attack of oxygen of the ester group onto the β-position of allene forms VI. Intermediate VI on demethylation leads to phosphinoylisocoumarin VII. This product VII further involves the double bond isomerization to lead to phosphinoyl isocoumarins 20, 21, 23, 26 and 27. The isomerization is not observed in the case of 22, 24, 25, 28 and 29. Alternatively, the cyclization may also proceed after the hydrolysis of ester group to –COOH due to the presence of adventitious moisture in trifluoroacetic acid.
![[1860-5397-10-99-i8]](/bjoc/content/inline/1860-5397-10-99-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Possible pathway for the formation of phosphinoylisocoumarins.
Scheme 8: Possible pathway for the formation of phosphinoylisocoumarins.
When the above reaction was performed in wet trifluoroacetic acid (TFA/H2O = 20:1) at 70 °C, phosphinoylisocoumarins were formed in all cases, but additionally, phosphorus-free isocoumarins 30–35 (Scheme 9) [37] are also formed in the reaction using terminally substituted allenes 4b–d and 4h–j. We have also determined the X-ray structure of compound 33 (Figure 5) for final confirmation. It is possible that isocoumarins 30–35 are formed via the intermediates VIII–IX (Scheme 10) [40]. The phosphorus moiety of IX may then be cleaved as Ph2POOH to form the phosphorus-free isocoumarins. Since this was not the interest in the present study, we did not proceed further.
![[1860-5397-10-99-i9]](/bjoc/content/inline/1860-5397-10-99-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reaction of allenes in wet trifluoroacetic acid.
Scheme 9: Reaction of allenes in wet trifluoroacetic acid.
![[1860-5397-10-99-5]](/bjoc/content/figures/1860-5397-10-99-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure of 33. Selected bond lengths [Å] with estimated standard deviations are given in parentheses: O2–C8 1.377(6), C8–C10 1.492(6).
Figure 5: Molecular structure of 33. Selected bond lengths [Å] with estimated standard deviations are given i...
![[1860-5397-10-99-i10]](/bjoc/content/inline/1860-5397-10-99-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Possible pathway for the formation of isocoumarins 30–35 (along with 21–23 and 27–29).
Scheme 10: Possible pathway for the formation of isocoumarins 30–35 (along with 21–23 and 27–29).
Conclusion
A fairly simple route to phosphinoylindoles and phosphinoylisocoumarins starting from functionalized propargyl alcohols via allenyl phosphine oxide is developed. The first reaction involves base-mediated deprotection and cyclization while the latter methodology involves acid mediation in which trifluoroacetic acid acts as the reagent as well as the solvent.
Experimental
Details on the synthesis of the compounds 1a–1m, 2a–2j, 3a–3c, 3m, 4a–4j and 5–35 are given in Supporing Information File 1.
Crystallographic data for the structures of 7, 9, 20 and 33 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 981067-981070. Copies of the data can be obtained free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(1223)336033 or e-mail: deposit@ccdc.cam.ac.uk]. The structures were solved and refined by standard methods [41-43].
7: Colorless block, C29H24NO2P, M = 449.46, monoclinic, space group P21/c, a = 9.9427(15), b = 16.894(3), c = 14.819(2) Å, α = 90.00, β = 109.195(2), γ = 90.00o, V = 2350.8(6) Å3, Z = 4, µ = 0.143 mm−1, data/restrains/parameters: 4141/0/299, R indices (I> 2σ(I)): R1 = 0.0408, wR2 (all data) = 0.1101. CCDC no. 981067.
9: Colorless block, C22H20NOP, M = 345.36, orthorhombic, space group Pccn, a = 11.2497(6), b = 21.1287(9), c = 15.2880(6) Å, α = 90, β = 90, γ = 90o, V = 3633.8(3) Å3, Z = 8, µ = 0.160 mm−1, data/restrains/parameters: 3205/0/231, R indices (I> 2σ(I)): R1 = 0.0430, wR2 (all data) = 0.1076. CCDC no. 981068.
20: Colorless block, C22H17O3P, M = 360.33, triclinic, space group ![[Graphic 1]](/bjoc/content/inline/1860-5397-10-99-i11.svg?max-width=637&scale=1.18182) , a = 9.7440(19), b = 9.9918(17), c = 10.2864(18) Å, α = 84.229(14), β = 76.556(16), γ = 66.323(18)o, V = 892.0(3) Å3, Z = 2, µ = 0.173 mm−1, data/restrains/parameters: 3647/0/236, R indices (I> 2σ(I)): R1 = 0.0455, wR2 (all data) = 0.1114. CCDC no. 981069.
, a = 9.7440(19), b = 9.9918(17), c = 10.2864(18) Å, α = 84.229(14), β = 76.556(16), γ = 66.323(18)o, V = 892.0(3) Å3, Z = 2, µ = 0.173 mm−1, data/restrains/parameters: 3647/0/236, R indices (I> 2σ(I)): R1 = 0.0455, wR2 (all data) = 0.1114. CCDC no. 981069.
33: Colorless needles, C11H9BrO2, M = 253.09, triclinic, space group , a = 7.9413(19), b = 7.9674(19), c = 9.746(2) Å, α = 66.05(2), β = 79.379(19), γ = 62.93(2)o, V = 501.8(2) Å3, Z = 2, µ = 4.064 mm−1, data/restrains/parameters: 1354/0/128, R indices (I> 2σ(I)): R1 = 0.0425, wR2 (all data) = 0.1021. CCDC no. 981070.
Supporting Information
| Supporting Information File 1: Details on the synthesis and characterization of the compounds 1a–1m, 2a–2j, 3a–3c, 3m, 4a–4j and 5–35 and 1H/13C NMR spectra of new compounds (including A–B). | ||
| Format: PDF | Size: 5.0 MB | Download |
| Supporting Information File 2: CIF file for the compounds 7, 9, 20 and 33. | ||
| Format: CIF | Size: 56.1 KB | Download |
Acknowledgements
We thank the Department of Science and Technology (DST, New Delhi) for financial support and the Single Crystal X-ray diffractometer facility, and the University Grants Commission (UGC, New Delhi) for equipment under UPE and CAS programs. G.G., R.K., M.N.R. thank the Council of Scientific and Industrial Research (CSIR, New Delhi) for fellowship. K.C.K.S. thanks DST for the J. C. Bose fellowship.
References
-
Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, 2004; pp 760–787.
Return to citation in text: [1] -
Brummond, K. M.; DeForrest, J. E. Synthesis 2007, 795. doi:10.1055/s-2007-965963
Return to citation in text: [1] -
Hoffmann-Röder, A.; Krause, N. Angew. Chem., Int. Ed. 2004, 43, 1196. doi:10.1002/anie.200300628
Return to citation in text: [1] [2] -
Rivera-Fuentes, P.; Diederich, F. Angew. Chem., Int. Ed. 2012, 51, 2818. doi:10.1002/anie.201108001
Return to citation in text: [1] -
Ma, S. Chem. Rev. 2005, 105, 2829. doi:10.1021/cr020024j
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Soc. Rev. 2010, 39, 783. doi:10.1039/b913749a
Return to citation in text: [1] -
Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074. doi:10.1002/anie.201101460
Return to citation in text: [1] -
Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994. doi:10.1021/cr1004088
Return to citation in text: [1] -
Pinho e Melo, T. M. V. D. Monatsh. Chem. 2011, 142, 681. doi:10.1007/s00706-011-0505-7
Return to citation in text: [1] -
Beccalli, E. M.; Bernasconi, A.; Borsini, E.; Broggini, G.; Rigamonti, M.; Zecchi, G. J. Org. Chem. 2010, 75, 6923. doi:10.1021/jo101501u
Return to citation in text: [1] -
Poonoth, M.; Krause, N. J. Org. Chem. 2011, 76, 1934. doi:10.1021/jo102416e
Return to citation in text: [1] -
Inuki, S.; Iwata, A.; Oishi, S.; Fujii, N.; Ohno, H. J. Org. Chem. 2011, 76, 2072. doi:10.1021/jo102388e
Return to citation in text: [1] -
Cheng, J.; Jiang, X.; Ma, S. Org. Lett. 2011, 13, 5200. doi:10.1021/ol202074e
Return to citation in text: [1] -
Szeto, J.; Sriramurthy, V.; Kwon, O. Org. Lett. 2011, 13, 5420. doi:10.1021/ol201730q
Return to citation in text: [1] -
Scheufler, F.; Maier, M. E. Eur. J. Org. Chem. 2000, 3945. doi:10.1002/1099-0690(200012)2000:23<3945::AID-EJOC3945>3.0.CO;2-6
Return to citation in text: [1] -
Jiang, X.; Kong, W.; Chen, J.; Ma, S. Org. Biomol. Chem. 2008, 6, 3606. doi:10.1039/b808767a
Return to citation in text: [1] -
Sajna, K. V.; Kotikalapudi, R.; Chakravarty, M.; Kumar, N. N. B.; Kumara Swamy, K. C. J. Org. Chem. 2011, 76, 920. doi:10.1021/jo102240u
Return to citation in text: [1] -
Moonen, K.; Laureyn, I.; Stevens, C. V. Chem. Rev. 2004, 104, 6177. doi:10.1021/cr030451c
Return to citation in text: [1] -
McGrath, J. W.; Chin, J. P.; Quinn, J. P. Nat. Rev. Microbiol. 2013, 11, 412. doi:10.1038/nrmicro3011
Return to citation in text: [1] -
Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029. doi:10.1021/cr020049i
Return to citation in text: [1] -
Chakravarty, M.; Kumara Swamy, K. C. J. Org. Chem. 2006, 71, 9128. doi:10.1021/jo061525y
Return to citation in text: [1] [2] -
Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 5345. doi:10.1021/jo300705f
Return to citation in text: [1] [2] [3] -
Chakravarty, M.; Bhuvan Kumar, N. N.; Sajna, K. V.; Kumara Swamy, K. C. Eur. J. Org. Chem. 2008, 4500. doi:10.1002/ejoc.200800490
Return to citation in text: [1] [2] -
Bhuvan Kumar, N. N.; Nagarjuna Reddy, M.; Kumara Swamy, K. C. J. Org. Chem. 2009, 74, 5395. doi:10.1021/jo900896v
Return to citation in text: [1] [2] [3] -
Phani Pavan, M.; Nagarjuna Reddy, M.; Bhuvan Kumar, N. N.; Kumara Swamy, K. C. Org. Biomol. Chem. 2012, 10, 8113. doi:10.1039/c2ob26285a
Return to citation in text: [1] [2] -
Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 8712. doi:10.1021/jo301694n
Return to citation in text: [1] [2] -
Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Tetrahedron Lett. 2011, 52, 5323. doi:10.1016/j.tetlet.2011.08.020
Return to citation in text: [1] [2] -
Rama Suresh, R.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 6959. doi:10.1021/jo301149s
Return to citation in text: [1] [2] -
Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Chem. Commun. 2011, 47, 5629. doi:10.1039/c1cc10230c
Return to citation in text: [1] [2] [3] [4] -
Phani Pavan, M.; Kumara Swamy, K. C. Synlett 2011, 1288. doi:10.1055/s-0030-1260533
Return to citation in text: [1] [2] -
Gangadhararao, G.; Kumara Swamy, K. C. Tetrahedron 2014, 70, 2643. doi:10.1016/j.tet.2014.02.064
Return to citation in text: [1] -
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi:10.1021/cr900211p
Return to citation in text: [1] -
Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620. doi:10.3390/molecules18066620
Return to citation in text: [1] -
Pal, S.; Chatare, V.; Pal, M. Curr. Org. Synth. 2011, 15, 782. doi:10.2174/138527211794518970
Return to citation in text: [1] -
Chincilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084. doi:10.1039/c1cs15071e
Return to citation in text: [1] -
Roesch, K. R.; Larock, R. C. J. Org. Chem. 2002, 67, 86. doi:10.1021/jo010579z
Return to citation in text: [1] -
Mikhailovskaya, T. F.; Vasilevsky, S. F. Russ. Chem. Bull. 2010, 59, 632. doi:10.1007/s11172-010-0133-0
Return to citation in text: [1] [2] -
Schuster, H. F.; Coppola, G. M. Allenes in Organic Synthesis; Wiley: New York, NY, 1984; pp 247 ff.
Return to citation in text: [1] -
Lang, R. W.; Hansen, H.-J. Organic Synthesis; Collect. Vol. 7; Wiley & Sons: New York, 1990; p 232.
Return to citation in text: [1] -
Kumara Swamy, K. C.; Satish Kumar, N. Acc. Chem. Res. 2006, 39, 324. doi:10.1021/ar050188x
(A review on pentacoordinated phosphorus).
Return to citation in text: [1] -
Sheldrick, G. M. SADABS, Siemens Area Detector Absorption Correction; University of Göttingen: Germany, 1996.
Return to citation in text: [1] -
Sheldrick, G. M. SHELX-97: A program for crystal structure solution and refinement; University of Göttingen: Germany, 1997.
Return to citation in text: [1] -
Sheldrick, G. M. SHELXTL NT Crystal Structure Analysis Package, Version 5; Bruker AXS Analytical X-ray System: WI (USA), 1999.
Return to citation in text: [1]
| 41. | Sheldrick, G. M. SADABS, Siemens Area Detector Absorption Correction; University of Göttingen: Germany, 1996. |
| 42. | Sheldrick, G. M. SHELX-97: A program for crystal structure solution and refinement; University of Göttingen: Germany, 1997. |
| 43. | Sheldrick, G. M. SHELXTL NT Crystal Structure Analysis Package, Version 5; Bruker AXS Analytical X-ray System: WI (USA), 1999. |
| 1. | Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, 2004; pp 760–787. |
| 2. | Brummond, K. M.; DeForrest, J. E. Synthesis 2007, 795. doi:10.1055/s-2007-965963 |
| 8. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994. doi:10.1021/cr1004088 |
| 9. | Pinho e Melo, T. M. V. D. Monatsh. Chem. 2011, 142, 681. doi:10.1007/s00706-011-0505-7 |
| 10. | Beccalli, E. M.; Bernasconi, A.; Borsini, E.; Broggini, G.; Rigamonti, M.; Zecchi, G. J. Org. Chem. 2010, 75, 6923. doi:10.1021/jo101501u |
| 11. | Poonoth, M.; Krause, N. J. Org. Chem. 2011, 76, 1934. doi:10.1021/jo102416e |
| 12. | Inuki, S.; Iwata, A.; Oishi, S.; Fujii, N.; Ohno, H. J. Org. Chem. 2011, 76, 2072. doi:10.1021/jo102388e |
| 13. | Cheng, J.; Jiang, X.; Ma, S. Org. Lett. 2011, 13, 5200. doi:10.1021/ol202074e |
| 14. | Szeto, J.; Sriramurthy, V.; Kwon, O. Org. Lett. 2011, 13, 5420. doi:10.1021/ol201730q |
| 28. | Rama Suresh, R.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 6959. doi:10.1021/jo301149s |
| 5. | Ma, S. Chem. Rev. 2005, 105, 2829. doi:10.1021/cr020024j |
| 6. | Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Soc. Rev. 2010, 39, 783. doi:10.1039/b913749a |
| 7. | Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074. doi:10.1002/anie.201101460 |
| 29. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Chem. Commun. 2011, 47, 5629. doi:10.1039/c1cc10230c |
| 4. | Rivera-Fuentes, P.; Diederich, F. Angew. Chem., Int. Ed. 2012, 51, 2818. doi:10.1002/anie.201108001 |
| 26. | Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 8712. doi:10.1021/jo301694n |
| 3. | Hoffmann-Röder, A.; Krause, N. Angew. Chem., Int. Ed. 2004, 43, 1196. doi:10.1002/anie.200300628 |
| 27. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Tetrahedron Lett. 2011, 52, 5323. doi:10.1016/j.tetlet.2011.08.020 |
| 21. | Chakravarty, M.; Kumara Swamy, K. C. J. Org. Chem. 2006, 71, 9128. doi:10.1021/jo061525y |
| 22. | Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 5345. doi:10.1021/jo300705f |
| 23. | Chakravarty, M.; Bhuvan Kumar, N. N.; Sajna, K. V.; Kumara Swamy, K. C. Eur. J. Org. Chem. 2008, 4500. doi:10.1002/ejoc.200800490 |
| 24. | Bhuvan Kumar, N. N.; Nagarjuna Reddy, M.; Kumara Swamy, K. C. J. Org. Chem. 2009, 74, 5395. doi:10.1021/jo900896v |
| 25. | Phani Pavan, M.; Nagarjuna Reddy, M.; Bhuvan Kumar, N. N.; Kumara Swamy, K. C. Org. Biomol. Chem. 2012, 10, 8113. doi:10.1039/c2ob26285a |
| 26. | Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 8712. doi:10.1021/jo301694n |
| 27. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Tetrahedron Lett. 2011, 52, 5323. doi:10.1016/j.tetlet.2011.08.020 |
| 28. | Rama Suresh, R.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 6959. doi:10.1021/jo301149s |
| 29. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Chem. Commun. 2011, 47, 5629. doi:10.1039/c1cc10230c |
| 30. | Phani Pavan, M.; Kumara Swamy, K. C. Synlett 2011, 1288. doi:10.1055/s-0030-1260533 |
| 23. | Chakravarty, M.; Bhuvan Kumar, N. N.; Sajna, K. V.; Kumara Swamy, K. C. Eur. J. Org. Chem. 2008, 4500. doi:10.1002/ejoc.200800490 |
| 24. | Bhuvan Kumar, N. N.; Nagarjuna Reddy, M.; Kumara Swamy, K. C. J. Org. Chem. 2009, 74, 5395. doi:10.1021/jo900896v |
| 25. | Phani Pavan, M.; Nagarjuna Reddy, M.; Bhuvan Kumar, N. N.; Kumara Swamy, K. C. Org. Biomol. Chem. 2012, 10, 8113. doi:10.1039/c2ob26285a |
| 3. | Hoffmann-Röder, A.; Krause, N. Angew. Chem., Int. Ed. 2004, 43, 1196. doi:10.1002/anie.200300628 |
| 18. | Moonen, K.; Laureyn, I.; Stevens, C. V. Chem. Rev. 2004, 104, 6177. doi:10.1021/cr030451c |
| 19. | McGrath, J. W.; Chin, J. P.; Quinn, J. P. Nat. Rev. Microbiol. 2013, 11, 412. doi:10.1038/nrmicro3011 |
| 15. | Scheufler, F.; Maier, M. E. Eur. J. Org. Chem. 2000, 3945. doi:10.1002/1099-0690(200012)2000:23<3945::AID-EJOC3945>3.0.CO;2-6 |
| 16. | Jiang, X.; Kong, W.; Chen, J.; Ma, S. Org. Biomol. Chem. 2008, 6, 3606. doi:10.1039/b808767a |
| 17. | Sajna, K. V.; Kotikalapudi, R.; Chakravarty, M.; Kumar, N. N. B.; Kumara Swamy, K. C. J. Org. Chem. 2011, 76, 920. doi:10.1021/jo102240u |
| 21. | Chakravarty, M.; Kumara Swamy, K. C. J. Org. Chem. 2006, 71, 9128. doi:10.1021/jo061525y |
| 22. | Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 5345. doi:10.1021/jo300705f |
| 29. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Chem. Commun. 2011, 47, 5629. doi:10.1039/c1cc10230c |
| 30. | Phani Pavan, M.; Kumara Swamy, K. C. Synlett 2011, 1288. doi:10.1055/s-0030-1260533 |
| 29. | Srinivas, V.; Sajna, K. V.; Kumara Swamy, K. C. Chem. Commun. 2011, 47, 5629. doi:10.1039/c1cc10230c |
| 37. | Mikhailovskaya, T. F.; Vasilevsky, S. F. Russ. Chem. Bull. 2010, 59, 632. doi:10.1007/s11172-010-0133-0 |
| 40. |
Kumara Swamy, K. C.; Satish Kumar, N. Acc. Chem. Res. 2006, 39, 324. doi:10.1021/ar050188x
(A review on pentacoordinated phosphorus). |
| 38. | Schuster, H. F.; Coppola, G. M. Allenes in Organic Synthesis; Wiley: New York, NY, 1984; pp 247 ff. |
| 39. | Lang, R. W.; Hansen, H.-J. Organic Synthesis; Collect. Vol. 7; Wiley & Sons: New York, 1990; p 232. |
| 24. | Bhuvan Kumar, N. N.; Nagarjuna Reddy, M.; Kumara Swamy, K. C. J. Org. Chem. 2009, 74, 5395. doi:10.1021/jo900896v |
| 32. | Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi:10.1021/cr900211p |
| 33. | Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620. doi:10.3390/molecules18066620 |
| 34. | Pal, S.; Chatare, V.; Pal, M. Curr. Org. Synth. 2011, 15, 782. doi:10.2174/138527211794518970 |
| 35. | Chincilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084. doi:10.1039/c1cs15071e |
| 36. | Roesch, K. R.; Larock, R. C. J. Org. Chem. 2002, 67, 86. doi:10.1021/jo010579z |
| 37. | Mikhailovskaya, T. F.; Vasilevsky, S. F. Russ. Chem. Bull. 2010, 59, 632. doi:10.1007/s11172-010-0133-0 |
| 31. | Gangadhararao, G.; Kumara Swamy, K. C. Tetrahedron 2014, 70, 2643. doi:10.1016/j.tet.2014.02.064 |
| 22. | Sajna, K. V.; Kumara Swamy, K. C. J. Org. Chem. 2012, 77, 5345. doi:10.1021/jo300705f |
© 2014 Gangadhararao et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)