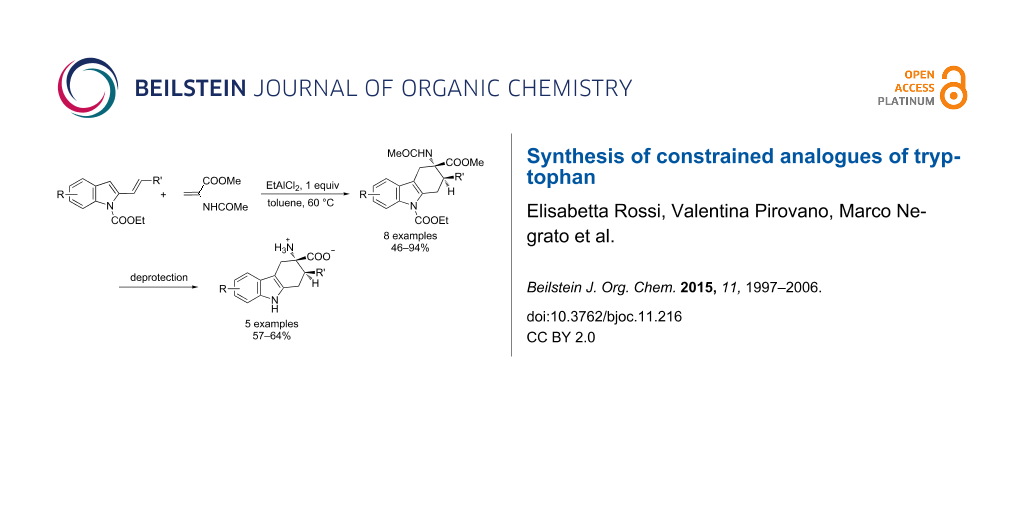
Abstract
A Lewis acid-catalysed diastereoselective [4 + 2] cycloaddition of vinylindoles and methyl 2-acetamidoacrylate, leading to methyl 3-acetamido-1,2,3,4-tetrahydrocarbazole-3-carboxylate derivatives, is described. Treatment of the obtained cycloadducts under hydrolytic conditions results in the preparation of a small library of compounds bearing the free amino acid function at C-3 and pertaining to the class of constrained tryptophan analogues.

Graphical Abstract
Introduction
With the term of “unnatural” amino acids, a plethora of naturally occurring or chemically synthesized non-proteinogenic amino acids are classified [1]. Chemically synthesized non-proteinogenic amino acids embody in principle a countless collection of assorted chemical structures and are mainly employed as they are or as scaffolds for pharmacologically active products and biochemical studies. In recent years, both pharmaceutical companies and academics became interested in the design and synthesis of peptidomimetics and peptide analogues as new therapeutic drugs [2,3]. Medicinal chemistry progress in these fields was probably inspired by the biochemical advancements in the recognition of new naturally occurring peptides possessing useful biological activities and in the elucidation of their physiological functions. However, peptides assembled with natural amino acids present several drawbacks related to metabolic instability, deficiency in selective interactions and reduced oral absorption that prevent their use in therapy [4]. On the other hand, peptidomimetics offer the advantage of nearly countless manipulations in order to control the biological functions, stability, potency, and ADME parameters [5]. In particular, the inclusion of the amino acidic framework in a cyclic or bicyclic structure confers specific features to the synthesized molecules: well-defined secondary structure, structural rigidity, enhanced binding activity and selectivity [6,7]. For example, bupivacaine (Figure 1), commercialized by Sanofi, is a local anesthetic drug containing a six-membered ring [8]. Moreover, fused bicyclic unnatural amino acids are present in the structures of two antiviral drugs, boceprevir (Merck) [9] and telaprevir (Vertex, Johnson & Johnson) [10] used against hepatitis C genotype 1 viral infections (Figure 1).
![[1860-5397-11-216-1]](/bjoc/content/figures/1860-5397-11-216-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of drugs embodying unnatural amino acids.
Figure 1: Examples of drugs embodying unnatural amino acids.
In this research field, tryptophan analogues received less attention with respect to others congeners. Constraints and modifications in the tryptophan core have been mainly attained following two different strategies: embodying the nitrogen atom of the amino acid function in a β-carboline framework or inserting a linking group between the α-carbon atom of the amino acid function and the C-2 carbon of the indole ring (tetrahydrocarbazole derivatives).
The first report on the synthesis and biological evaluation of a constrained tryptophan analogue appeared in the literature in 1973 [11]. Maki and co-workers reported the synthesis of 3-amino-1,2,3,4-tetrahydrocarbazole-3-carboxylic acid as a rigid analogue of α-methyltryptophan, a well known unnatural amino acid able to inhibit α-chymotrypsin activity, Figure 2 (A). Hardening tryptophan in β-carboline or carbazole frameworks has been used by Hénichart and co-workers in their studies devoted to the identification of new dual NK1/NK2 antagonists [12], as shown in Figure 2 (B).
In 2003, the pharmaceutical company Zentaris patented a series of tetrahydrocarbazole derivatives as ligands for G-protein-coupled receptors (GPCR), and in particular as antagonists of the gonadotropin-releasing hormone (GnRH) [13,14], Figure 2 (C). Finally, in the field of therapeutic peptides, constrained tryptophan residues of the β-carboline family (L-Tpi and D-Tpi), were used by Grieco and co-workers in the study and development of urotensin-II receptor (UTR) peptide ligands [15], Figure 2 (D).
![[1860-5397-11-216-2]](/bjoc/content/figures/1860-5397-11-216-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Examples of biologically active compounds embodying constrained analogues of tryptophan.
Figure 2: Examples of biologically active compounds embodying constrained analogues of tryptophan.
Switching from the biological evaluation to the chemistry of tryptophan analogues, the above described two categories were synthesized according to the Pictet–Spengler reaction or by Fischer indole synthesis [11-14]. However, Fischer indolization suffers from the lack of regioselectivity depending on the substitution pattern of starting materials (substituted arylhydrazines and cyclohexanones) and is therefore useful mainly for the synthesis of symmetrically or unsubstituted derivatives [16,17].
Recently, our research group described the synthesis of tetrahydrocarbazole derivatives by Diels–Alder reactions between 2-vinylindoles as 4π-components with activated dienophiles and allenes [18-21]. The reported methodologies allowed for the synthesis of substituted derivatives with excellent degrees of selectivity. Starting from these results, we envisaged that 3-amino-1,2,3,4-tetrahydrocarbazole-3-carboxylic acid derivatives 3, constrained analogues of tryptophan, could be synthesized by Diels–Alder reactions between 2-vinylindoles 1 [22] as dienes and methyl 2-acetamidoacrylate (dehydroalanine) 2 [23] as dienophile (Scheme 1).
![[1860-5397-11-216-i1]](/bjoc/content/inline/1860-5397-11-216-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Planned Diels–Alder reactions for the synthesis of tetrahydrocarbazoles as constrained analogues of tryptophan.
Scheme 1: Planned Diels–Alder reactions for the synthesis of tetrahydrocarbazoles as constrained analogues of...
Reported [4 + 2] cycloaddition reactions of methyl 2-acetamidoacrylate (2) and its congeners with cyclic/acyclic dienes and azadienes occur under conventional heating or microwave irradiation [24]. Moreover, the use of titanium tetrachloride as Lewis acidic promoter has been reported [25]. Finally, simple functionalization reactions of indoles with 2 are reported in the literature [26-29].
In this paper we report our findings about the reactions between 2-vinylindoles 1 and methyl 2-acetamidoacrylate (2) resulting in the synthesis of a small library of 3-amino-1,2,3,4-tetrahydrocarbazole-3-carboxylic acid derivatives 3. Moreover, a study was focused on the complete deprotection of the cycloadducts in order to obtain the free amino acid function.
Results
2-Vinylindole 1a was selected as a benchmark substrate to evaluate the feasibility of the devised [4 + 2] cycloaddition reaction with methyl 2-acetamidoacrylate (2). Test conditions and obtained results are summarized in Table 1.
Table 1: Screening of reaction conditions for the cycloaddition reaction between 1a and 2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-216-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Catalyst | mol % | Solvent | T, °C | Time, h | Yield, % | dr 3a:3’a |
|---|---|---|---|---|---|---|---|
| 1 | – | – | toluene | 110 | 24 | NR | – |
| 2 | Mg(ClO4)2 | 15 | toluene | 110 | 24 | NRb | – |
| 3 | Sc(OTf)3 | 15 | toluene | 110 | 24 | NRb | – |
| 4 | Sc(OTf)3 | 15 | CHCl3 | 40 | 24 | NRb | – |
| 5 | Cu(OTf)2 | 30 | CHCl3 | 60 | 24 | NRc | – |
| 6 | BF3·OEt2 | 15 | toluene | rt, 100 | 48 | NRb | – |
| 7 | BF3·OEt2 | 50 | toluene | rt, 100 | 48 | NRb | – |
| 8 | AuCl3 | 5 | toluene | rt, 100 | 24 | NRb | – |
| 9 | Au(PPh3)Cl/AgOTf | 2 | toluene | rt, 100 | 22 | 20b | 1:1 |
| 10 | EtAlCl2 | 20 | CHCl3 | rt | 48 | 35b | >98:2 |
| 11 | EtAlCl2 | 20 | toluene | rt | 48 | 30b | >98:2 |
| 12 | EtAlCl2 | 100 | CHCl3 | rt | 48 | 57 | >98:2 |
| 13 | EtAlCl2 | 100 | CHCl3 | 60 | 3 | 83 | >98:2 |
| 14 | EtAlCl2 | 100 | toluene | 60 | 5 | 94 | >98:2 |
aReaction conditions: A solution of 2 (0.22 mmol) and the catalyst in the appropriate solvent (2 mL, 0.1 M) was stirred at room temperature for 1 h, then 1a (0.2 mmol) was added and the mixture stirred at the stated time and temperature. bStarting materials recovered at the end of the reaction. cMixture of unidentified compounds.
The reaction was ineffective under thermal conditions (Table 1, entry 1). As a consequence we tested simple Lewis acids as potential promoters for the transformation. Magnesium perchlorate, scandium and copper triflate and boron trifluoride failed to give the desired compounds (Table 1, entries 2–7). Switching the reaction medium from toluene to chloroform (Table 1, entry 4), or increasing the catalyst loading, entry 7, were also ineffective. In all reactions tested the starting materials were recovered unreacted at the end of the reaction; decomposition was observed only using copper(II) triflate as catalyst (Table 1, entry 5). Unsatisfactory results were obtained also in the presence of gold(III) and gold(I) catalysts (Table 1, entries 8 and 9). Only in the presence of a cationic gold(I) complex the diastereoisomeric cycloadducts 3a and 3'a were isolated in 20% overall yield and in a diastereomeric ratio of 1:1 (Table 1, entry 9). These results were quite surprising as these catalyst/solvent systems were effective in our previously reported Diels–Alder cycloadditions involving 1a as diene [18-21]. In particular, under gold catalysis excellent results in term of yields and selectivity were achieved [20,21]. We next turned our attention to aluminium catalysis. The use of 20 mol % of ethylaluminium dichloride in chloroform or toluene at room temperature resulted in the isolation of 3a and 3'a in 35% and 30% overall yield, respectively, in excellent diastereoisomeric ratios, higher than 98:2 in favour of the 2,3-trans adduct (Table 1, entries 10 and 11). Better yields could be obtained increasing the catalyst loading to 100 mol % (Table 1, entry 12) whereas the best results were achieved working under the same reaction conditions at 60 °C, in chloroform or toluene as solvents (Table 1, entries 13 and 14). Under these conditions 3a and 3'a were isolated in 83% and 94% overall yields, respectively, preserving the same diastereoisomeric ratios. With the best reaction conditions in hands, the scope of the transformation was then explored using the 2-vinylindoles 1a–j. Results are shown in Table 2.
Table 2: Scope of the cycloaddition reactions between 2-vinylindoles 1a–j and methyl 2-acetamidoacrylate (2).
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-216-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | 2-Vinylindole | Solvent | T, °C | Time, h | Products | Yield, % | dr 3:3' |
|---|---|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-216-i7.svg?max-width=637&scale=1.0)
1a |
CHCl3 | 60 | 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-216-i8.svg?max-width=637&scale=1.0)
(±)-3a |
83 | >98:2 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-216-i9.svg?max-width=637&scale=1.0)
1a |
toluene | 60 | 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-216-i10.svg?max-width=637&scale=1.0)
(±)-3a |
94 | >98:2 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-216-i11.svg?max-width=637&scale=1.0)
1b |
CHCl3 | 60 | 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-216-i12.svg?max-width=637&scale=1.0)
(±)-3b, (±)-3'b |
86a | 1:1 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-216-i13.svg?max-width=637&scale=1.0)
1b |
toluene | 60 | 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-216-i14.svg?max-width=637&scale=1.0)
(±)-3b |
84 | >98:2 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-216-i15.svg?max-width=637&scale=1.0)
1c |
toluene | 60 | 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-216-i16.svg?max-width=637&scale=1.0)
(±)-3c |
74 | >98:2 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-216-i17.svg?max-width=637&scale=1.0)
1d |
toluene | 60 | 4 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-216-i18.svg?max-width=637&scale=1.0)
(±)-3d |
83 | >98:2 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-216-i19.svg?max-width=637&scale=1.0)
1e |
toluene | 60 | 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-216-i20.svg?max-width=637&scale=1.0)
(±)-3e |
79 | >98:2 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-216-i21.svg?max-width=637&scale=1.0)
1f |
toluene | 60 | 5 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-216-i22.svg?max-width=637&scale=1.0)
(±)-3f |
78 | >98:2 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-216-i23.svg?max-width=637&scale=1.0)
1g |
toluene | 60 | 24 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-216-i24.svg?max-width=637&scale=1.0)
(±)-3g |
50 | >98:2 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-216-i25.svg?max-width=637&scale=1.0)
1h |
toluene | 60 | 24 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-216-i26.svg?max-width=637&scale=1.0)
(±)-3h |
46 | >98:2 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-216-i27.svg?max-width=637&scale=1.0)
1i |
toluene | 60 | 5 | – | –b | – |
| 12 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-216-i28.svg?max-width=637&scale=1.0)
1i |
toluene | rt | 5 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-216-i29.svg?max-width=637&scale=1.0)
4 |
33 | – |
| 13 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-216-i30.svg?max-width=637&scale=1.0)
1j |
toluene | rt | 5 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-216-i31.svg?max-width=637&scale=1.0)
(±)-3i |
44 | – |
| 14 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-216-i32.svg?max-width=637&scale=1.0)
1j |
CHCl3 | rt | 5 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-216-i33.svg?max-width=637&scale=1.0)
(±)-3i |
12 | – |
aOverall yield (3 + 3’), pure isolated compounds after chromatographic purification. bMixture of unidentified compounds.
Table 2, entries 1 and 2 report the best results for the cycloaddition reaction of 1a with 2, obtained during the reaction conditions screening (see Table 1). Quite surprisingly, indole 1b bearing a methyl group at the distal position of the diene system, gives rise to the desired cycloadducts 3b and 3'b in good yields but without any diastereoselectivity when we performed the reaction in chloroform (Table 2, entry 3). Nevertheless, switching from chloroform to toluene as reaction solvent, results in a total regain of diastereoselectivity without loss of efficiency (Table 2, entry 4). As a consequence, we choose toluene as solvent for further cycloaddition reactions. Under these conditions, alkyl- (Table 2, entries 5 and 6) and aryl- (entries 7 and 8) substituted dienes smoothly react with methyl 2-acetamidoacrylate (2) affording the desired cycloadducts in good yields and excellent diastereoselectivities. A considerable drop in yield is observed using dienes substituted in position 5 of the indole ring with an EWG or an EDG such as fluorine or methoxy (Table 2, entries 9 and 10). Finally, the indole 1i, unsubstituted at the terminal position of the diene system, decomposes at 60 °C (Table 2, entry 11) whereas working at room temperature, entry 12, the main isolated compound, beside unreacted 1i and 2, is the vinylindole dimer 4 (33% yield) arising from the cycloaddition reaction between two molecules of vinylindole 1i. However, the same reaction performed with the N-unsubstituted vinylindole 1j in toluene or chloroform as solvents and at room temperature (Table 2, entries 13 and 14) allows for the isolation of the desired cycloadduct 3i, in moderate and poor yields, respectively. A small quantity of a dimeric compound analogous to 4 was observed in the crude reaction mixture, via 1H NMR, along with a mixture of unidentified compounds.
The diastereoisomeric mixtures (3/3') could be easily separated by flash chromatography and pure isomers were characterized by combined one- (1H NMR, 13C NMR-APT) and two-dimensional (COSY, HMBC, NOESY) experiments, performed at 300 MHz, using C6D6 or CDCl3 as solvents. In particular, the regiochemistry and stereochemistry around the C1/C4 moiety were assigned on the basis of spatial coupling interactions detected by 2D-NOESY experiments. As an example, the regiochemistry of the Diels–Alder adducts 3a and 3'a was reasonably assigned on the basis of NOE interactions between H-5 and the two H-4 hydrogens, Figure 3. Furthermore, for compound 3a it was possible to observe diagnostic NOE interactions between NH and the three cis hydrogens in position 1, 2 and 4, Figure 3. On the contrary, for compound 3'a in Figure 3, NH NOE interactions involve cis hydrogens at positions 1 and 4 and the aromatic p-tolyl hydrogens, Figure 3.
![[1860-5397-11-216-3]](/bjoc/content/figures/1860-5397-11-216-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Structure elucidation of diastereoisomeric tetrahydrocarbazoles 3a and 3’a via NMR experiments.
Figure 3: Structure elucidation of diastereoisomeric tetrahydrocarbazoles 3a and 3’a via NMR experiments.
Finally, tetrahydrocarbazoles 3a,b,d and 3'a were selected as substrates to test the reactivity of our compounds under hydrolytic conditions to obtain the deprotection of indole nitrogen and the free amino acid function at C-3, Scheme 2. The deprotection of indole nitrogen, giving rise to compounds 5a–d, was achieved in high yields in the presence of 1 equiv of potassium carbonate, in methanol at reflux for 2 h. After characterisation, the obtained compounds 5a–d and 3i were treated first with hydrochloric acid (12 N) in a multimode microwave oven at 120 °C for 2 h, then with an excess of propylene oxide in ethanol at reflux for 1 h, and finally purified by flash chromatography. The whole reaction sequence was realized in a one-flask procedure. The obtained compounds 6a–e and the relative reaction yields, referred to the starting compounds 3/3', are reported in Scheme 2.
![[1860-5397-11-216-i2]](/bjoc/content/inline/1860-5397-11-216-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of unprotected tryptophan derivatives 6a–e.
Scheme 2: Synthesis of unprotected tryptophan derivatives 6a–e.
No epimerisation reactions occurred during the hydrolytic processes as demonstrated via 2D NMR experiments (COSY, HSQC).
Discussion
In the Diels–Alder reaction of vinylindoles 1 with methyl 2-acetamidoacrylate (2), among tested Lewis acid, only EtAlCl2 is able to trigger the reaction toward the formation of the desired cycloadducts 3/3'. Similarly, Piersanti and co-workers reported the unique capability of EtAlCl2, with respect to related hard Lewis acids, to activate 2 toward the nucleophilic addition of indoles [29]. They ascribed the observed reactivity to the formation of a complex between EtAlCl2 and 2, verified via 1H NMR experiments, and involving coordination with both amide and ester carbonyl groups [29]. Conceivably, the same activated complex participated in our Diels–Alder reactions, see footnote a in Table 1. Moreover, the high diastereoselectivity observed with indoles 1a–h in toluene as solvent, accounts for a concerted or a pseudoconcerted mechanism in which the formation of an endo transition state and thus of the final 2,3-trans adducts 3a–h is preferred (Scheme 3).
![[1860-5397-11-216-i3]](/bjoc/content/inline/1860-5397-11-216-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible reaction mechanism for the cycloaddition reactions of indoles 1a–h with 2 in toluene.
Scheme 3: Plausible reaction mechanism for the cycloaddition reactions of indoles 1a–h with 2 in toluene.
Thus bond formation between the 1,4-carbon terminus of the diene and the double bond of the dienophile occurs almost simultaneously allowing for the preservation of the stereochemistry around the outer-ring diene double bond. Besides, in the presence of a terminal p-tolyl group on the diene moiety (see Table 2, entries 1 and 2) the diastereoselectivity is not affected by the reaction medium (chloroform vs toluene). Moving to the alkyl-substituted vinylindole 1b, the reaction yields equimolecular amounts of both conceivable diastereoisomers in CHCl3 whereas toluene is the optimal solvent to achieve high diastereoselection (see Table 2, entries 3 and 4). It is well known that reaction rates and selectivity in Diels–Alder reactions are affected by solvents [30-32]. In particular, polarity and hydrogen bond donor ability of the solvent can impact the diastereoisomeric ratios in a Diels–Alder reaction. Such an effect seems negligible in the reaction of 1a with 2 and effective in the reaction of 1b with 2. In this latter case, the reaction outcome could be explained by the formation of two energetically comparable transition states (endo + exo TS) or by the occurrence of a stepwise mechanism. Finally, the results obtained with indole 1i, unsubstituted at the terminal position of the diene system, can be attributed to the thermal instability of 1i under standard reaction conditions and to the lack of reactivity toward 2 at room temperature. The lack of reactivity of 1i toward 2 can be overcome working with N-unprotected indole 1j. Modulations in reactivity upon specific substitution patterns at the indole nucleus were observed by us in competitive Diels–Alder cycloaddition/Michael addition reactions of vinylindoles with classical dienophile and in competitive cycloaddition/hydroarylation reactions with allenes, see [20,21] for a more detailed discussion. Conceivably, removal of the EWG at the nitrogen modifies the electronic distribution of the reacting diene allowing the desired reaction to take place. Moreover, a model reaction performed with the 2-(4-methylstyryl)-1H-indole (1k), unprotected at N1 and comparable to 1a at the diene moiety, and 2 afforded the corresponding tetrahydrocarbazoles 5a and 5b in 1:1.3 diasteroisomeric ratio (Scheme 4).
![[1860-5397-11-216-i4]](/bjoc/content/inline/1860-5397-11-216-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cycloaddition reaction of 2-vinylindole 1k and methyl 2-acetamidoacrylate (2).
Scheme 4: Cycloaddition reaction of 2-vinylindole 1k and methyl 2-acetamidoacrylate (2).
As reported for the reaction between 1b and 2 in chloroform, the reaction outcome could be explained by the formation of two energetically comparable transition states (endo + exo TS) or by the occurrence of a stepwise mechanism. Distinction between the two proposed mechanisms cannot be made on the basis of experimental evidences. Therefore, isolation or detection in the reaction mixtures of plausible intermediates were unsuccessful and a computational study devoted to the identification of the most suitable transition state is beyond the scope of this work.
Conclusion
In summary, we reported a flexible approach to the synthesis of an important class of compounds like constrained tryptophan derivatives, pertinent to the class of tetrahydrocabazoles. The synthesis was realised through an intermolecular [4 + 2]-cycloaddition of 2-vinylindoles and methyl 2-acetamidoacrylate. Although the reaction requires a stoichiometric amount of EtAlCl2 as promoter, it presents several advantages with respect to classical Fischer indole synthesis, normally adopted for the preparation of these derivatives [11-13]. First of all, modulation of substituents around the terahydrocarbazole nucleus is achievable without the formation of regioisomeric derivatives. Moreover, there is no need of multistep sequences for the synthesis of starting materials. Methyl 2-acetamidoacrylate is a commercially available and cheap reactant, whereas 2-vinylindoles can be easily synthesized by a common precursor [22]. Finally, by exploring the scope of the reaction and in connection with our previous reports on the cycloaddition reactions of 2-vinylindoles, we were able to point out several features about the reactivity of these compounds. In particular, the dependence upon the substitution pattern at nitrogen and at the outer-ring double bond, highlighted the need to select the appropriate promoter for each desired transformation.
Supporting Information
| Supporting Information File 1: Experimental procedures and analytical data. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Lu, Y.; Freeland, S. Genome Biol. 2006, 7, No. 102. doi:10.1186/gb-2006-7-1-102
Return to citation in text: [1] -
Craik, D. J.; Fairlie, D. P.; Liras, S.; Price, D. Chem. Biol. Drug Des. 2013, 81, 136–147. doi:10.1111/cbdd.12055
Return to citation in text: [1] -
Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Chem. Rev. 2014, 114, 901–926. doi:10.1021/cr400031z
Return to citation in text: [1] -
Vagner, J.; Qu, H.; Hruby, V. J. Curr. Opin. Chem. Biol. 2008, 12, 292–296. doi:10.1016/j.cbpa.2008.03.009
Return to citation in text: [1] -
Liskamp, R. M. J.; Rijkers, D. T. S.; Kruijtzer, J. A. W.; Kemmink, J. ChemBioChem 2011, 12, 1626–1653. doi:10.1002/cbic.201000717
Return to citation in text: [1] -
Hanessian, S.; Auzzas, L. Acc. Chem. Res. 2008, 41, 1241–1251. doi:10.1021/ar8000052
Return to citation in text: [1] -
Stevenazzi, A.; Marchini, M.; Sandrone, G.; Vergani, B.; Lattanzio, M. Bioorg. Med. Chem. Lett. 2014, 24, 5349–5356. doi:10.1016/j.bmcl.2014.10.016
Return to citation in text: [1] -
Adger, B.; Dyer, U.; Hutton, G.; Woods, M. Tetrahedron Lett. 1996, 37, 6399–6402. doi:10.1016/0040-4039(96)01357-3
Return to citation in text: [1] -
Rotella, D. P. Expert Opin. Drug Discovery 2013, 8, 1439–1447. doi:10.1517/17460441.2013.843525
Return to citation in text: [1] -
Kwong, A. D.; Kauffman, R. S.; Hurter, P.; Mueller, P. Nat. Biotechnol. 2011, 29, 993–1003. doi:10.1038/nbt.2020
Return to citation in text: [1] -
Maki, Y.; Masugi, T.; Hiramitsu, T.; Ogiso, T. Chem. Pharm. Bull. 1973, 21, 2460–2465. doi:10.1248/cpb.21.2460
Return to citation in text: [1] [2] [3] -
Millet, R.; Goossens, J.-F.; Bertrand-Caumont, K.; Chavatte, P.; Houssin, R.; Hénichart, J.-P. Lett. Pept. Sci. 1999, 6, 221–233. doi:10.1023/A:1008844323931
Return to citation in text: [1] [2] [3] -
Koppitz, M. K.; Muhn, H. P.; Shaw, K. J.; Hess-Stumpp, H.; Paulini, K. W. Tetrahydrocarbazol derivatives as ligands for G-protein-coupled receptors (GPCR). U.S. Patent US 2003/0232873 A1, Dec 18, 2003.
Return to citation in text: [1] [2] [3] -
Koppitz, M.; Reinhardt, G.; van Lingen, A. Tetrahedron Lett. 2005, 46, 911–914. doi:10.1016/j.tetlet.2004.12.058
Return to citation in text: [1] [2] -
Carotenuto, A.; Auriemma, L.; Merlino, F.; Limatola, A.; Campiglia, P.; Gomez-Monterrey, I.; d’Emmanuele di Villa Bianca, R.; Brancaccio, D.; Santicioli, P.; Meini, S.; Maggi, C. A.; Novellino, E.; Grieco, P. J. Pept. Sci. 2013, 19, 293–300. doi:10.1002/psc.2498
Return to citation in text: [1] -
Trofimov, B. A.; Nedolya, N. A. In Comprehensive Heterocyclic Chemistry; Jones, G.; Ramsden, C. A., Eds.; Elsevier: Oxford, U.K., 2008; Vol. 3, pp 88–168.
Return to citation in text: [1] -
Bender, M.; Christoffers, J. Z. Naturforsch. 2011, 66b, 1209–1218.
Return to citation in text: [1] -
Abbiati, G.; Canevari, V.; Facoetti, D.; Rossi, E. Eur. J. Org. Chem. 2007, 517–525. doi:10.1002/ejoc.200600625
Return to citation in text: [1] [2] -
Pirovano, V.; Abbiati, G.; Dell’Acqua, M.; Facoetti, D.; Giordano, M.; Rossi, E. Synlett 2012, 23, 2913–2918. doi:10.1055/s-0032-1317588
Return to citation in text: [1] [2] -
Pirovano, V.; Dell’Acqua, M.; Facoetti, D.; Nava, D.; Rizzato, S.; Abbiati, G.; Rossi, E. Eur. J. Org. Chem. 2013, 6267–6279. doi:10.1002/ejoc.201300725
Return to citation in text: [1] [2] [3] [4] -
Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594–3596. doi:10.1039/c3cc41514g
Return to citation in text: [1] [2] [3] [4] -
Rossi, E.; Abbiati, G.; Canevari, V.; Celentano, G.; Magri, E. Synthesis 2006, 299–304. doi:10.1055/s-2005-918509
Return to citation in text: [1] [2] -
Crestey, F.; Collot, V.; Stiebing, S.; Rault, S. Synthesis 2006, 3506–3514. doi:10.1055/s-2006-950242
Return to citation in text: [1] -
Kotha, S.; Bandarugattu, V. B.; Krishna, N. G. Tetrahedron 2014, 70, 5361–5384. doi:10.1016/j.tet.2014.05.056
Return to citation in text: [1] -
Avenoza, A.; Cativiela, C.; Fernández-Recio, M. A.; Peregrina, J. M. Synlett 1995, 891–892. doi:10.1055/s-1995-5125
Return to citation in text: [1] -
Mari, M.; Lucarini, S.; Bartoccini, F.; Piersanti, G.; Spadoni, G. Beilstein J. Org. Chem. 2014, 10, 1991–1998. doi:10.3762/bjoc.10.207
Return to citation in text: [1] -
Lucarini, S.; Mari, M.; Piersanti, G.; Spadoni, G. RSC Adv. 2013, 3, 19135–19143. doi:10.1039/c3ra42922a
Return to citation in text: [1] -
Pirovano, V.; Facoetti, D.; Dell’Acqua, M.; Della Fontana, E.; Abbiati, G.; Rossi, E. Org. Lett. 2013, 15, 3812–3815. doi:10.1021/ol401716b
Return to citation in text: [1] -
Angelini, E.; Balsamini, C.; Bartoccini, F.; Lucarini, S.; Piersanti, G. J. Org. Chem. 2008, 73, 5654–5657. doi:10.1021/jo800881u
Return to citation in text: [1] [2] [3] -
Gholami, M. R.; Talebi, B. A. J. Phys. Org. Chem. 2003, 16, 369–372. doi:10.1002/poc.647
Return to citation in text: [1] -
Moreau, Y.; Loos, P.-F.; Assfeld, X. Theor. Chem. Acc. 2004, 112, 228–239. doi:10.1007/s00214-004-0581-4
Return to citation in text: [1] -
García Ruano, J. L.; Clemente, F. R.; González Gutiérrez, L.; Gordillo, R.; Martín Castro, A. M.; Rodríguez Ramos, J. H. J. Org. Chem. 2002, 67, 2926–2933. doi:10.1021/jo016175o
Return to citation in text: [1]
| 22. | Rossi, E.; Abbiati, G.; Canevari, V.; Celentano, G.; Magri, E. Synthesis 2006, 299–304. doi:10.1055/s-2005-918509 |
| 1. | Lu, Y.; Freeland, S. Genome Biol. 2006, 7, No. 102. doi:10.1186/gb-2006-7-1-102 |
| 6. | Hanessian, S.; Auzzas, L. Acc. Chem. Res. 2008, 41, 1241–1251. doi:10.1021/ar8000052 |
| 7. | Stevenazzi, A.; Marchini, M.; Sandrone, G.; Vergani, B.; Lattanzio, M. Bioorg. Med. Chem. Lett. 2014, 24, 5349–5356. doi:10.1016/j.bmcl.2014.10.016 |
| 18. | Abbiati, G.; Canevari, V.; Facoetti, D.; Rossi, E. Eur. J. Org. Chem. 2007, 517–525. doi:10.1002/ejoc.200600625 |
| 19. | Pirovano, V.; Abbiati, G.; Dell’Acqua, M.; Facoetti, D.; Giordano, M.; Rossi, E. Synlett 2012, 23, 2913–2918. doi:10.1055/s-0032-1317588 |
| 20. | Pirovano, V.; Dell’Acqua, M.; Facoetti, D.; Nava, D.; Rizzato, S.; Abbiati, G.; Rossi, E. Eur. J. Org. Chem. 2013, 6267–6279. doi:10.1002/ejoc.201300725 |
| 21. | Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594–3596. doi:10.1039/c3cc41514g |
| 5. | Liskamp, R. M. J.; Rijkers, D. T. S.; Kruijtzer, J. A. W.; Kemmink, J. ChemBioChem 2011, 12, 1626–1653. doi:10.1002/cbic.201000717 |
| 22. | Rossi, E.; Abbiati, G.; Canevari, V.; Celentano, G.; Magri, E. Synthesis 2006, 299–304. doi:10.1055/s-2005-918509 |
| 4. | Vagner, J.; Qu, H.; Hruby, V. J. Curr. Opin. Chem. Biol. 2008, 12, 292–296. doi:10.1016/j.cbpa.2008.03.009 |
| 11. | Maki, Y.; Masugi, T.; Hiramitsu, T.; Ogiso, T. Chem. Pharm. Bull. 1973, 21, 2460–2465. doi:10.1248/cpb.21.2460 |
| 12. | Millet, R.; Goossens, J.-F.; Bertrand-Caumont, K.; Chavatte, P.; Houssin, R.; Hénichart, J.-P. Lett. Pept. Sci. 1999, 6, 221–233. doi:10.1023/A:1008844323931 |
| 13. | Koppitz, M. K.; Muhn, H. P.; Shaw, K. J.; Hess-Stumpp, H.; Paulini, K. W. Tetrahydrocarbazol derivatives as ligands for G-protein-coupled receptors (GPCR). U.S. Patent US 2003/0232873 A1, Dec 18, 2003. |
| 14. | Koppitz, M.; Reinhardt, G.; van Lingen, A. Tetrahedron Lett. 2005, 46, 911–914. doi:10.1016/j.tetlet.2004.12.058 |
| 2. | Craik, D. J.; Fairlie, D. P.; Liras, S.; Price, D. Chem. Biol. Drug Des. 2013, 81, 136–147. doi:10.1111/cbdd.12055 |
| 3. | Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Chem. Rev. 2014, 114, 901–926. doi:10.1021/cr400031z |
| 16. | Trofimov, B. A.; Nedolya, N. A. In Comprehensive Heterocyclic Chemistry; Jones, G.; Ramsden, C. A., Eds.; Elsevier: Oxford, U.K., 2008; Vol. 3, pp 88–168. |
| 17. | Bender, M.; Christoffers, J. Z. Naturforsch. 2011, 66b, 1209–1218. |
| 11. | Maki, Y.; Masugi, T.; Hiramitsu, T.; Ogiso, T. Chem. Pharm. Bull. 1973, 21, 2460–2465. doi:10.1248/cpb.21.2460 |
| 13. | Koppitz, M. K.; Muhn, H. P.; Shaw, K. J.; Hess-Stumpp, H.; Paulini, K. W. Tetrahydrocarbazol derivatives as ligands for G-protein-coupled receptors (GPCR). U.S. Patent US 2003/0232873 A1, Dec 18, 2003. |
| 14. | Koppitz, M.; Reinhardt, G.; van Lingen, A. Tetrahedron Lett. 2005, 46, 911–914. doi:10.1016/j.tetlet.2004.12.058 |
| 10. | Kwong, A. D.; Kauffman, R. S.; Hurter, P.; Mueller, P. Nat. Biotechnol. 2011, 29, 993–1003. doi:10.1038/nbt.2020 |
| 15. | Carotenuto, A.; Auriemma, L.; Merlino, F.; Limatola, A.; Campiglia, P.; Gomez-Monterrey, I.; d’Emmanuele di Villa Bianca, R.; Brancaccio, D.; Santicioli, P.; Meini, S.; Maggi, C. A.; Novellino, E.; Grieco, P. J. Pept. Sci. 2013, 19, 293–300. doi:10.1002/psc.2498 |
| 9. | Rotella, D. P. Expert Opin. Drug Discovery 2013, 8, 1439–1447. doi:10.1517/17460441.2013.843525 |
| 8. | Adger, B.; Dyer, U.; Hutton, G.; Woods, M. Tetrahedron Lett. 1996, 37, 6399–6402. doi:10.1016/0040-4039(96)01357-3 |
| 12. | Millet, R.; Goossens, J.-F.; Bertrand-Caumont, K.; Chavatte, P.; Houssin, R.; Hénichart, J.-P. Lett. Pept. Sci. 1999, 6, 221–233. doi:10.1023/A:1008844323931 |
| 25. | Avenoza, A.; Cativiela, C.; Fernández-Recio, M. A.; Peregrina, J. M. Synlett 1995, 891–892. doi:10.1055/s-1995-5125 |
| 23. | Crestey, F.; Collot, V.; Stiebing, S.; Rault, S. Synthesis 2006, 3506–3514. doi:10.1055/s-2006-950242 |
| 24. | Kotha, S.; Bandarugattu, V. B.; Krishna, N. G. Tetrahedron 2014, 70, 5361–5384. doi:10.1016/j.tet.2014.05.056 |
| 20. | Pirovano, V.; Dell’Acqua, M.; Facoetti, D.; Nava, D.; Rizzato, S.; Abbiati, G.; Rossi, E. Eur. J. Org. Chem. 2013, 6267–6279. doi:10.1002/ejoc.201300725 |
| 21. | Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594–3596. doi:10.1039/c3cc41514g |
| 11. | Maki, Y.; Masugi, T.; Hiramitsu, T.; Ogiso, T. Chem. Pharm. Bull. 1973, 21, 2460–2465. doi:10.1248/cpb.21.2460 |
| 12. | Millet, R.; Goossens, J.-F.; Bertrand-Caumont, K.; Chavatte, P.; Houssin, R.; Hénichart, J.-P. Lett. Pept. Sci. 1999, 6, 221–233. doi:10.1023/A:1008844323931 |
| 13. | Koppitz, M. K.; Muhn, H. P.; Shaw, K. J.; Hess-Stumpp, H.; Paulini, K. W. Tetrahydrocarbazol derivatives as ligands for G-protein-coupled receptors (GPCR). U.S. Patent US 2003/0232873 A1, Dec 18, 2003. |
| 29. | Angelini, E.; Balsamini, C.; Bartoccini, F.; Lucarini, S.; Piersanti, G. J. Org. Chem. 2008, 73, 5654–5657. doi:10.1021/jo800881u |
| 30. | Gholami, M. R.; Talebi, B. A. J. Phys. Org. Chem. 2003, 16, 369–372. doi:10.1002/poc.647 |
| 31. | Moreau, Y.; Loos, P.-F.; Assfeld, X. Theor. Chem. Acc. 2004, 112, 228–239. doi:10.1007/s00214-004-0581-4 |
| 32. | García Ruano, J. L.; Clemente, F. R.; González Gutiérrez, L.; Gordillo, R.; Martín Castro, A. M.; Rodríguez Ramos, J. H. J. Org. Chem. 2002, 67, 2926–2933. doi:10.1021/jo016175o |
| 20. | Pirovano, V.; Dell’Acqua, M.; Facoetti, D.; Nava, D.; Rizzato, S.; Abbiati, G.; Rossi, E. Eur. J. Org. Chem. 2013, 6267–6279. doi:10.1002/ejoc.201300725 |
| 21. | Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594–3596. doi:10.1039/c3cc41514g |
| 29. | Angelini, E.; Balsamini, C.; Bartoccini, F.; Lucarini, S.; Piersanti, G. J. Org. Chem. 2008, 73, 5654–5657. doi:10.1021/jo800881u |
| 26. | Mari, M.; Lucarini, S.; Bartoccini, F.; Piersanti, G.; Spadoni, G. Beilstein J. Org. Chem. 2014, 10, 1991–1998. doi:10.3762/bjoc.10.207 |
| 27. | Lucarini, S.; Mari, M.; Piersanti, G.; Spadoni, G. RSC Adv. 2013, 3, 19135–19143. doi:10.1039/c3ra42922a |
| 28. | Pirovano, V.; Facoetti, D.; Dell’Acqua, M.; Della Fontana, E.; Abbiati, G.; Rossi, E. Org. Lett. 2013, 15, 3812–3815. doi:10.1021/ol401716b |
| 29. | Angelini, E.; Balsamini, C.; Bartoccini, F.; Lucarini, S.; Piersanti, G. J. Org. Chem. 2008, 73, 5654–5657. doi:10.1021/jo800881u |
| 18. | Abbiati, G.; Canevari, V.; Facoetti, D.; Rossi, E. Eur. J. Org. Chem. 2007, 517–525. doi:10.1002/ejoc.200600625 |
| 19. | Pirovano, V.; Abbiati, G.; Dell’Acqua, M.; Facoetti, D.; Giordano, M.; Rossi, E. Synlett 2012, 23, 2913–2918. doi:10.1055/s-0032-1317588 |
| 20. | Pirovano, V.; Dell’Acqua, M.; Facoetti, D.; Nava, D.; Rizzato, S.; Abbiati, G.; Rossi, E. Eur. J. Org. Chem. 2013, 6267–6279. doi:10.1002/ejoc.201300725 |
| 21. | Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594–3596. doi:10.1039/c3cc41514g |
© 2015 Rossi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)