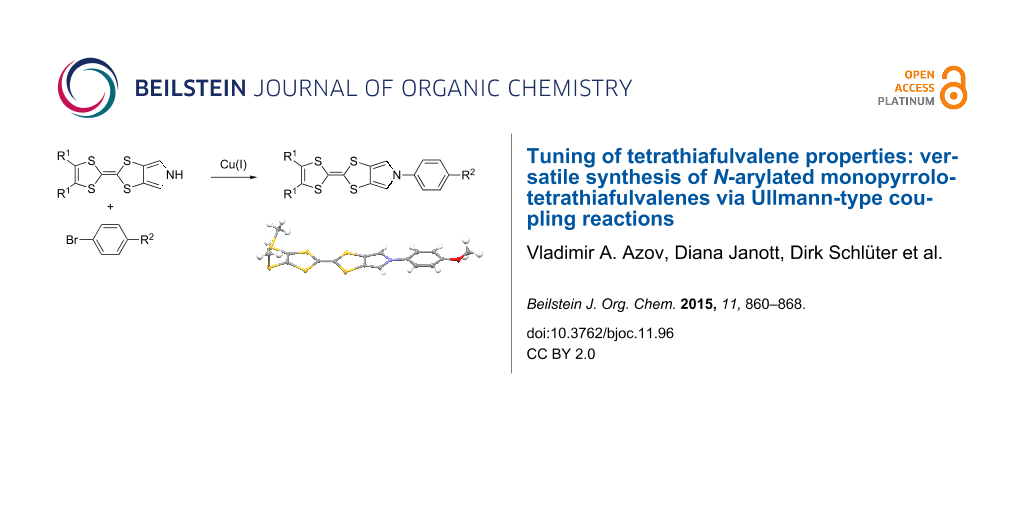
Abstract
An Ullmann-type coupling reaction was employed for the preparation of several N-arylated monopyrrolotetrathiafulvalenes with variable substitution patterns. Spectroscopic and electrochemical properties of the coupling products strongly depend on the electronic nature of the aromatic substituents due to their direct conjugation with the tetrathiafulvalene chromophore. The crystal packing of the arylated monopyrrolotetrathiafulvalenes is primarily defined by networks of C–H···X weak hydrogen bonds and short S···S contacts involving the tetrathiafulvalene moieties.

Graphical Abstract
Introduction
For the last four decades tetrathiafulvalenes [1,2] (Figure 1) 1 have been the subject of extensive studies due to their outstanding electron-donating properties and ability to induce reversible electrochemically-induced switching processes in molecular and supramolecular systems [3,4]. Availability of selective synthetic methods [5,6] gave access to differently substituted tetrathiafulvalene (TTF) moieties which allowed tuning of oxidation potential, donating ability, as well as other physical and chemical properties. The regioselective functionalization of TTF, however, remains problematic due to the presence of four identical attachment sites. Incorporation of the TTF moiety in macrocycles usually leads to poorly separable mixtures of cis/trans isomers [7-9]. Even if separation is possible, TTFs are prone to cis/trans isomerization, which can be induced by light [10] or traces of acid [11]. These problems are aggravated by the fact that each reversible oxidation–reduction cycle of the TTF moiety always leads to formation of cis/trans isomer mixtures.
![[1860-5397-11-96-1]](/bjoc/content/figures/1860-5397-11-96-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structures of tetrathiafulvalenes 1, bis-pyrrolotetrathiafulvalenes 2 and monopyrrolotetrathiafulvalenes 3.
Figure 1: Molecular structures of tetrathiafulvalenes 1, bis-pyrrolotetrathiafulvalenes 2 and monopyrrolotetr...
Bis-pyrrolotetrathiafulvalenes 2 and monopyrrolotetrathiafulvalenes (MPTTFs) 3 represent a significant modification of the TTF backbone featuring a more extended electron-rich π-system with only two or three easily accessible attachment points for external substituents, respectively [12,13]. The asymmetric nature of MPTTFs 3 opens the possibility for the introduction of different R- and R1-groups on the two sides of the TTF moiety. If R1 = alkyl (or a similar group with an sp3-hybridized carbon atom), such substituents can be readily attached to the pre-formed MPTTF moiety using a variety of common nucleophilic substitution reactions. In the case of R1 = aryl, two possible approaches for the preparation of N-arylated MPTTF derivatives 4 have been reported (Scheme 1). In the first procedure [14-16], the aryl substituent is incorporated during the initial synthetic steps to form a N-aryl-1,3-dithiolo[4,5-c]pyrrole-2-thione 5, which is then coupled to 1,3-dithiole-2-thione 6 in hot triethyl or trimethyl phosphite. Using the second approach [17-19], the aryl group is attached to the MPTTF moiety using a direct copper-mediated Ullmann-type N-arylation reaction [20,21]; this method was also used for the preparation of arylated bis-pyrrolotetrathiafulvalenes 2. Although being reported in the literature, it has so far not found wide spread use and was employed only with a narrow scope of aromatic derivatives with electron-donating substituents and alkylthio-substituted (R = SAlkyl) MPTTFS.
![[1860-5397-11-96-i1]](/bjoc/content/inline/1860-5397-11-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The two synthetic approaches used for the preparation of arylated monopyrrolotetrathiafulvalenes 1.
Scheme 1: The two synthetic approaches used for the preparation of arylated monopyrrolotetrathiafulvalenes 1.
Being interested in the preparation of calix[4]arene receptors with MPTTF moieties directly attached to an aromatic calixarene backbone, we have chosen the copper-catalysed N-arylation reaction as a method for coupling of aromatic and MPTTF moieties with each other and successfully employed it for the preparation of two bis-MPTTF-calixarene conjugates, as well as two model low molecular weight aromatic derivatives 4a and 4c [22]. To explore the scope of the reaction, we decided to conduct a deeper investigation varying reaction conditions and testing different substituents on the aromatic as well as the MPTTF components of the reaction. It led to the preparation of a family of MPTTF aromatic derivatives, whose properties and crystal structures are discussed below.
Results and Discussion
Synthesis
Our initial synthetic efforts were focused on optimizing reaction conditions using the PrS-MPTTF derivative 7a [23] and bromoanisole 8a as starting materials (Scheme 2), as well as comparing two possible copper(I) ligands (Figure 2): trans-diaminocyclohexane (DACH) 9a, which was employed before in N-arylations with pyrrolo-TTFs [17-19], and its Schiff base derivative 9b, which was reported to be one of the most effective ligands in similar N-arylation reactions with other substrates [20,21].
![[1860-5397-11-96-i2]](/bjoc/content/inline/1860-5397-11-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of arylated monopyrrolotetrathiafulvalenes 4a–f.
Scheme 2: Synthesis of arylated monopyrrolotetrathiafulvalenes 4a–f.
![[1860-5397-11-96-2]](/bjoc/content/figures/1860-5397-11-96-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Initial experiments gave evidence of much lower efficiency of ligand 9b in comparison to 9a in terms of conversion and presence of undesired side-products. Reaction utilizing trans-diaminocyclohexane 9aalso allowed for fast optimization and reproducibility, which prompted us to focus our attention on the CuI/9a catalysis system. Although copper(I) is supposed to play a catalytic role [20,21], smaller CuI loadings led to diminished reaction yields, supposedly due to lower solubility/activity of the CuBr formed in the coupling reaction. Taking into account the affordability of CuI, its excess cannot be considered a disadvantage of the method. Additionally, use of the inexpensive Cu(I) catalyst allows to avoid Buchwald–Hartwig amination [24,25], which employs more expensive Pd-based catalysts for a similar type of C–N coupling reactions.
In a typical procedure, 1 equiv of MPTTF 7a, 7b [12], or 7c [26], 1.5–1.6 equiv of a brominated aromatic derivative, 0.5 equiv of CuI/9a and 3–4 equiv of K3PO4 were heated at 110–115 °C overnight in absolute dioxane in a Schlenk tube. The reaction yields amounted to 70–80% for stable MPTTF derivatives 4a,b,d,e, but were lower for 4c due to sensitivity of the starting material 7b, as well as for 4f due to its tendency towards oxidation.
Thus, the N-arylation reaction can be readily employed with electron-rich as well as electron-deficient aromatic derivatives, as well as with thioalkyl-substituted and non-substituted MPTTFs. The successful reaction with bromophenol 8d to form the adduct 4f also confirmed the possibility of the reaction with hydroxy-substituted aryl derivatives, paving the way for application of this method with non-protected calix[4]arene derivatives [27].
Compounds 4a–c,e,f (Figure 3) display UV–vis spectra typical for TTF derivatives with absorption maxima at λmax of ca. 310–330 nm as well as a long tail with very low absorption spanning to ca. 500 nm and rendering the yellow colour to the compounds. Thioalkyl TTF derivatives 4a,b,e,f also show a sloping shoulder at ca. 390 nm, which is missing in the non-substituted 4c. In contrast, the spectrum of nitro-derivative 4d displays an additional strong absorption band centred at ca. 425 nm, arising most likely due to charge transfer from the electron-rich MPTTF moiety to the electron-deficient aromatic substituent. This absorption manifests itself in the dark red colour of 4d, both in solid state as well as in solution.
![[1860-5397-11-96-3]](/bjoc/content/figures/1860-5397-11-96-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of compounds 4a,c–f (CH2Cl2, c = 4 × 10−5 M).
Figure 3: UV–vis spectra of compounds 4a,c–f (CH2Cl2, c = 4 × 10−5 M).
MPTTF derivatives are readily soluble in non-polar organic solvents, such as CH2Cl2, toluene or acetone (with the exception of poorly soluble 4c), giving solutions that are stable at room temperature in air.
Electrochemistry
Solution oxidation potentials of aromatic MPTTF conjugates were determined using cyclic voltammetry (CV) in CH2Cl2/Bu4NClO4 solution and are summarized in Table 1. The CVs of all compounds displayed two reversible oxidation waves on the cathodic scan (Figure 4) characteristic to TTFs [1], the first one leading to the radical cation and the second to the dication. Non-substituted derivative 4c shows a lower first oxidation potential than its alkylS-substituted counterpart 4a,b, as expected due to the electron-withdrawing effect of the two thioalkyl groups [28,29]. The strong electron-withdrawing effect of the 4-nitrophenyl group in 4d manifests itself in an increased oxidation potential with a shift of ca. 0.08 V for both oxidation waves. Aromatic electron-donating groups as in 4e and 4f do barely influence the potential of the two oxidation waves. Instead, they induce an additional oxidation wave at higher potentials of 1.06 V and 1.77 V, in 4e and 4f (see Figure S7, Supporting Information File 1), respectively. For the phenol derivative 4f, this oxidation is irreversible.
Table 1: Electrochemical data.a
aData were obtained using a one-compartment cell in CH2Cl2/0.1 M Bu4NClO4, Pt as the working and counter electrodes and a non-aqueous Ag/Ag+ reference electrode; scan rate 100 mV/s. Values given at room temperature vs SCE; the Fc/Fc+ couple (0.480 V vs SCE) was used as an internal reference [30].
![[1860-5397-11-96-4]](/bjoc/content/figures/1860-5397-11-96-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Cyclic voltammograms of compounds 4a,c [22] and 4d–f (plotted vs SCE; CH2Cl2/0.1 M Bu4NClO4).
Figure 4: Cyclic voltammograms of compounds 4a,c [22] and 4d–f (plotted vs SCE; CH2Cl2/0.1 M Bu4NClO4).
Crystal structures
Compounds 4a,b,d,e afforded high quality crystals that could be analysed using X-ray crystallography, allowing to unambiguously confirm the identity of the compounds and analyse their structural properties and arrangement in the solid state. Bond lengths and angles in all structures may be considered normal. With the exception of alkylS-substituents, the molecular frameworks of 4a,b,d,e display relatively low deviations from planarity. Angles between the least-square planes, defined by the heavy atoms of the aromatic ring and neighbouring pyrrole ring do not exceed 17.3° (in 4e, see Table 2), ensuring good conjugation between the MPTTF and aromatic moieties. Boat-type deviations of the TTF groups (folding along the S–S vectors in the five-membered rings) are minor for electron-deficient 4d lying below 5°, whereas they are much larger in electron-rich derivatives, where they reach 20.45° in 4a, 13.58° in 4b and 11.23° in 4e. This observation corresponds well with previously reported data: the electron-deficient N-Ts derivative has an almost planar arrangement of the TTF moiety, whereas N-alkyl derivatives show significant deviation from planarity [12].
Interestingly, the crystal structures of all four compounds feature similar packing arrangements with two crystallographically distinct molecules (Z’ = 2) with quasi-parallel tilted edge-to face arrangements. In the crystal packing, molecules are interconnected by multiple non-classical weak intermolecular hydrogen bonds [31] and C–H···π and S···S interactions, the latter being common in the crystals of sulfur-rich compounds such as TTFs [32]. Most of the close contacts involve the central parts of neighbouring molecules, thus connecting them with each other and leading to formation of supramolecular layers. Parallel layers are only loosely bound to each other [33]. This layered arrangement manifests itself in the crystal morphology: all Ar-MPTTF derivatives crystallize in the form of thin platelets, with the plane of the parallel molecular layers coinciding with the direction normal to the largest crystal face of the platelets. This observation can be rationalized by assuming fast crystal growth within each supramolecular layer, assisted by the presence of directed weak hydrogen bonds as well as C–H···π and S···S interactions. Addition of new parallel layers, connected to the previous layers via dispersive interactions of the van der Waals type, can be assumed to be a much slower process, leading to plate like crystals that mimic the layered makeup at the molecular level. Crystals of 4d, for example, form plates with an aspect ratio of 10 and above. Such layered arrangements make these derivatives possible candidates for organic electronics [34] and may serve as a motivation for evaluation of their electronic properties in the solid state.
Figures 5–8 display some aspects of molecular packing of MPTTF derivatives 4a,b,d,e.
![[1860-5397-11-96-5]](/bjoc/content/figures/1860-5397-11-96-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Crystal packing of 4a viewed along the a axis and showing one layer of molecules. Short S···S contacts are shown as black and S···C contacts as green dashed lines. Only the major orientation of the disordered propyl chain is shown.
Figure 5: Crystal packing of 4a viewed along the a axis and showing one layer of molecules. Short S···S conta...
![[1860-5397-11-96-6]](/bjoc/content/figures/1860-5397-11-96-6.png?scale=1.72&max-width=1024&background=FFFFFF)
Figure 6: Crystal packing of 4b showing a group of four molecules interconnected by multiple weak hydrogen bonds, C–H···π, and S···S close contacts.
Figure 6: Crystal packing of 4b showing a group of four molecules interconnected by multiple weak hydrogen bo...
![[1860-5397-11-96-7]](/bjoc/content/figures/1860-5397-11-96-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Crystal packing of 4d viewed along the b axis. Molecules of 4d form layers parallel to the (001) plane being interconnected with each other by means of short S···S contacts (green) and C–H···O2N weak hydrogen bonds (blue).
Figure 7: Crystal packing of 4d viewed along the b axis. Molecules of 4d form layers parallel to the (001) pl...
![[1860-5397-11-96-8]](/bjoc/content/figures/1860-5397-11-96-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Crystal packing of 4e showing a group of four molecules interconnected by multiple weak hydrogen bonds, C–H···π, and S···S close contacts.
Figure 8: Crystal packing of 4e showing a group of four molecules interconnected by multiple weak hydrogen bo...
Conclusion
In summary, several arylated monopyrrolotetrathiafulvalene derivatives have been conveniently prepared using a copper-mediated Ullmann-type N-arylation reaction. The reaction was shown to tolerate substituents of different nature on the aromatic group, as well as can be employed with substituted and non-substituted MPTTFs, opening the way for its application for the synthesis of more complex molecular systems, such as conjugates with non-protected calixarenes. New aromatic-MPTTF conjugates were characterized using different analytical methods and X-ray crystallography.
Experimental
Copper-catalyzed N-arylation reaction of monopyrrolo-tetrathiafulvalenes, general procedure. The reaction was performed in a similar manner as described before [22]. A heavy walled Schlenk tube with a wide bore Teflon screw stopcock was charged with MPTTF derivatives 7a, 7b or 7c, CuI, K3PO4, (±)-trans-1,2-diaminocyclohexane and an aromatic bromide, then absolute dioxane was added via syringe. The reaction mixture was degassed by three freeze-pump-thaw cycles, the vessel was filled with nitrogen, tightly sealed and stirred at 110–115 °C. The reaction was complete in 18–24 h (TLC control). The solvent was removed under reduced pressure directly from the Schlenk tube, the residue was dissolved in CH2Cl2, filtered through a plug of celite, and evaporated to dryness. The crude products were triturated with n-hexane to remove the unreacted aromatic starting material and then purified by flash chromatography on silica gel to afford pure N-arylated MPTTFs.
Preparation and characterization of 2-[4,5-bis(propylthio)-1,3-dithiol-2-ylidene]-5-(4-methoxyphenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4a) and 2-[1,3-dithiol-2-ylidene]-5-(4-methoxyphenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4c) was described before [22], also including the X-ray data of 4a (see Supporting Information File 1 and Supporting Information File 2). For 4a, corrected oxidation potentials are reported in Table 1.
2-[4,5-Bis(methylthio)-1,3-dithiol-2-ylidene]-5-(4-methoxyphenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4b). Prepared from 7b (0.040 g, 0.119 mmol), CuI (0.023 g, 0.119 mmol), K3PO4 (0.020 g, 0.954 mmol), trans-diaminocyclohexane (22 µL, 0.179 mmol) and 4-bromoanisole (8a, 0.033 g, 0.179 mmol) in 3 mL of dry dioxane. The product was purified by flash chromatography (CH2Cl2/cyclohexane, 1:2) to afford bright yellow crystals. X-ray quality crystals were grown by slow evaporation of CDCl3 solution. Yield: 43.2 mg (0.098 mmol, 82%). Mp 198–200 °C; Rf = 0.32 (CH2Cl2/cyclohexane, 1:2); 1H NMR (360 MHz, CDCl3) δ 7.25–7.20 (m, 2H), 6.95–6.91 (m, 2H), 6.79 (s, 2H), 3.83 (s, 3H), 2.43 (s, 6H); 13C NMR (90 MHz, CDCl3) δ 158.0, 134.0, 127.1, 121.9, 121.2, 120.2, 114.7, 111.4, 111.1, 55.6, 19.2; UV–vis (CH2Cl2) λmax (ε): 309 nm (25600 L∙mol−1∙cm−1), 329 (24500); MS (EI) m/z (%): 441 (100) [M]+•, 426 (10) [M − Me]+; HRMS (EI) m/z: [M]+• calcd for C17H15NOS6+•, 440.94780; found, 440.94675; CV (vs SCE, CH2Cl2): E1/2ox1 = 0.48 V, E1/2ox2 = 0.83 V.
2-[4,5-Bis(propylthio)-1,3-dithiol-2-ylidene]-5-(4-nitrophenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4d). Prepared from 7a (0.055 g, 0.140 mmol), CuI (0.014 g, 0.070 mmol), K3PO4 (0.089 g, 0.42 mmol), trans-diaminocyclohexane (7.5 µL, 0.062 mmol) and 4-bromonitrobenzene (8b, 0.045 g, 0.224 mmol) in 2 mL of dry dioxane. The product was purified by flash chromatography (CH2Cl2) to afford deep red crystals. X-ray quality crystals were grown by slow evaporation of CDCl3/heptane solution. Yield: 59.7 mg (0.116 mmol, 83%). Mp 240–244 °C; Rf = 0.72 (CH2Cl2); 1H NMR (360 MHz, CDCl3) δ 8.33–8.29 (m, 2H), 7.44–7.40 (m, 2H), 6.99 (s, 2H), 2.81 (t, 3J = 7.2 Hz, 4H), 1.68 (sext, 3J = 7.2 Hz, 4H), 1.02 (t, 3J = 7.2 Hz, 6H); 13C NMR (90 MHz, CDCl3) δ 144.7, 144.2, 127.5, 125.8, 125.3, 118.6, 117.3, 113.7, 110.1, 38.2, 23.1, 13.2; UV–vis (CH2Cl2) λmax (ε): 325 nm (25600 L∙mol−1∙cm−1), 426 (11200); MS (EI) m/z (%): 512 (100) [M]+•, 469 (5) [M − Pr]+∙, 436 (20) [M − HSPr]+•; HRMS (EI) m/z: [M]+• calcd for C20H20N2O2S6+•, 511.98435; found, 511.98296; CV (vs SCE, CH2Cl2): E1/2ox1 = 0.55 V, E1/2ox2 = 0.92 V.
2-[4,5-Bis(propylthio)-1,3-dithiol-2-ylidene]-5-(4-dimethylaminophenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4e). Prepared from 7a (0.050 g, 0.128 mmol), CuI (0.012 g, 0.063 mmol), K3PO4 (0.082 g, 0.386 mmol), trans-diaminocyclohexane (7.5 µL, 0.062 mmol) and 4-iodo-N,N-dimethylaniline (8c, 0.050 g, 0.202 mmol) in 2 mL of dry dioxane. The product was purified by flash chromatography (CH2Cl2/cyclohexane, 1:1) to afford light orange crystals. X-ray quality crystals were grown by slow evaporation of CDCl3/heptane solution. Yield: 53.8 mg (0.105 mmol, 82%); Mp 196–199 °C; Rf = 0.5 (CH2Cl2/cyclohexane, 1:1); 1H NMR (360 MHz, CD2Cl2) δ 7.22–7.17 (m, 2H), 6.81 (s, 2H), 6.76–6.71 (m, 2H), 2.96 (s, 6H), 2.81 (t, 3J = 7.2 Hz, 4H), 1.66 (sext, 3J = 7.2 Hz, 4H), 1.01 (t, 3J = 7.2 Hz, 6H); 13C NMR (90 MHz, CD2Cl2) δ 149.6, 130.7, 127.9, 122.0, 120.6, 119.8, 113.1, 111.9, 111.1, 40.8, 38.5, 23.5, 13.2; UV–vis (CH2Cl2) λmax (ε): 314 nm (34000 L∙mol−1∙cm−1), 330 (33600); MS (EI) m/z (%): 510 (100) [M]+•, 434 (20) [M − HSPr]+•; HRMS (EI) m/z: [M]+• calcd. for C22H26N2S6+•, 510.04147; found, 510.04104; CV (vs SCE, CH2Cl2): E1/2ox1 = 0.46 V, E1/2ox2 = 0.84 V, E1/2ox3 = 1.06 V.
2-[4,5-Bis(propylthio)-1,3-dithiol-2-ylidene]-5-(4-hydroxyphenyl)-5H-1,3-dithiolo[4,5-c]pyrrole (4f). Prepared from 7a (0.049 g, 0.125 mmol), CuI (0.012 g, 0.063 mmol), K3PO4 (0.082 g, 0.386 mmol), trans-diaminocyclohexane (7.5 µL, 0.062 mmol) and 4-bromophenol (8d, 0.035 g, 0.202 mmol) in 2 mL of dry dioxane. The product was purified by flash chromatography (CH2Cl2) to afford yellow crystals. Yield: 5 mg (0.052 mmol, 42%); Mp 123–125 °C; Rf = 0.27 (CH2Cl2); 1H NMR (200 MHz, CDCl3) δ 7.21–7.14 (m, 2H), 6.91–6.83 (m, 2H), 6.77 (s, 2H), 4.91 (s, 1H), 2.81 (t, 3J = 7.2 Hz, 4H), 1.66 (sext, 3J = 7.2 Hz, 4H), 1.01 (t, 3J = 7.2 Hz, 6H); 13C NMR (50 MHz, CDCl3) δ 153.9, 134.1, 129.7, 127.5, 122.2, 121.4, 119.2, 116.2, 111.4, 38.2, 23.1, 13.2; UV–vis (CH2Cl2) λmax (ε): 309 nm (26000 L∙mol−1∙cm−1), 326 (25900); MS (EI) m/z (%): 483 (100) [M]+•, 440 (5) [M − Pr]+∙, 407 (25) [M −HSPr]+•; HRMS (EI) m/z: [M]+• calcd. for C20H20N2O2S6+•, 482.99419; found, 482.99474; CV (vs SCE, CH2Cl2): E1/2ox1 = 0.47 V, E1/2ox2 = 0.86 V, E1/2ox3 = 1.77 V.
Supporting Information
| Supporting Information File 1: Experimental details, details on electrochemical characterization, 1H and 13C NMR spectra of compounds 4b,d–f, UV–vis spectrum and CV of 4b, as well as full crystal structure descriptions. | ||
| Format: PDF | Size: 1.4 MB | Download |
| Supporting Information File 2: Zip archive containing X-ray crystallographic data for 4a (CCDC 987551), 4b (CCDC 1049639), 4d (CCDC 1049638) and 4e (CCDC 1049637). | ||
| Format: ZIP | Size: 1.3 MB | Download |
Acknowledgements
We are grateful to Dr. T. Dülcks, Ms. D. Kemken (MS) and Ms. Ziyan Wang (NMR) for their help with the characterization of new compounds, and Dr. Arunpatcha Nimthong-Roldán for collection of X-ray data and structure refinement for compound 4b. The X-ray diffractometers were funded by NSF Grants 0087210 and 1337296, Ohio Board of Regents Grant CAP-491, and by Youngstown State University.
References
-
Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::AID-ANIE1372>3.0.CO;2-I
Return to citation in text: [1] [2] -
Yamada, J.; Sugimoto, T. TTF Chemistry. Fundamentals and Applications of Tetrathiafulvalene; Springer: Heidelberg, Germany, 2004.
Return to citation in text: [1] -
Becher, J.; Jeppesen, J. O.; Nielsen, K. Synth. Met. 2003, 133–134, 309–315. doi:10.1016/S0379-6779(02)00379-X
Return to citation in text: [1] -
Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n
Return to citation in text: [1] -
Fabre, J. M. Chem. Rev. 2004, 104, 5133–5150. doi:10.1021/cr0306440
Return to citation in text: [1] -
Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485
Return to citation in text: [1] -
Li, Z.-T.; Stein, P. C.; Becher, J.; Jensen, D.; Mørk, P.; Svenstrup, N. Chem. – Eur. J. 1996, 2, 624–633. doi:10.1002/chem.19960020605
Return to citation in text: [1] -
Le Derf, F.; Mazari, M.; Mercier, N.; Levillain, E.; Trippé, G.; Riou, A.; Richomme, P.; Becher, J.; Garín, J.; Orduna, J.; Gallego-Planas, N.; Gorgues, A.; Sallé, M. Chem. – Eur. J. 2001, 7, 447–455. doi:10.1002/1521-3765(20010119)7:2<447::AID-CHEM447>3.0.CO;2-A
Return to citation in text: [1] -
Azov, V. A.; Cordes, J.; Schlüter, D.; Dülcks, T.; Böckmann, M.; Doltsinis, N. L. J. Org. Chem. 2014, 79, 11714–11721. doi:10.1021/jo502469z
Return to citation in text: [1] -
Ballardini, R.; Balzani, V.; Becher, J.; Di Fabio, A.; Gandolfi, M. T.; Mattersteig, G.; Nielsen, M. B.; Raymo, F. M.; Rowan, S. J.; Stoddart, J. F.; White, A. J. P.; Williams, D. J. J. Org. Chem. 2000, 65, 4120–4126. doi:10.1021/jo0001941
Return to citation in text: [1] -
Souizi, A.; Robert, A.; Batail, P.; Ouahab, L. J. Org. Chem. 1987, 52, 1610–1611. doi:10.1021/jo00384a044
Return to citation in text: [1] -
Jeppesen, J. O.; Takimiya, K.; Jensen, F.; Brimert, T.; Nielsen, K.; Thorup, N.; Becher, J. J. Org. Chem. 2000, 65, 5794–5805. doi:10.1021/jo000742a
Return to citation in text: [1] [2] [3] -
Jeppesen, J. O.; Becher, J. Eur. J. Org. Chem. 2003, 3245–3266. doi:10.1002/ejoc.200300078
Return to citation in text: [1] -
Yin, B.; Yang, Y.; Cong, Z.; Imafuku, K. Heterocycles 2004, 63, 1577–1584. doi:10.3987/COM-04-10083
Return to citation in text: [1] -
Balandier, J.-Y.; Chas, M.; Dron, P. I.; Goeb, S.; Canevet, D.; Belyasmine, A.; Allain, M.; Sallé, M. J. Org. Chem. 2010, 75, 1589–1599. doi:10.1021/jo902529e
Return to citation in text: [1] -
Nygaard, S.; Leung, K. C.-F.; Aprahamian, I.; Ikeda, T.; Saha, S.; Laursen, B. W.; Kim, S.-Y.; Hansen, S. W.; Stein, P. C.; Flood, A. H.; Stoddart, J. F.; Jeppesen, J. O. J. Am. Chem. Soc. 2007, 129, 960–970. doi:10.1021/ja0663529
Return to citation in text: [1] -
Li, H.; Lambert, C. Chem. – Eur. J. 2006, 12, 1144–1155. doi:10.1002/chem.200500928
Return to citation in text: [1] [2] -
Li, J.; Zhang, G.; Zhang, D.; Zheng, R.; Shi, Q.; Zhu, D. J. Org. Chem. 2010, 75, 5330–5333. doi:10.1021/jo1007306
Return to citation in text: [1] [2] -
Solano, M. V.; DellaPia, E. A.; Jevric, M.; Schubert, C.; Wang, X.; van der Pol, C.; Kadziola, A.; Nørgaard, K.; Guldi, D. M.; Nielsen, M. B.; Jeppesen, J. O. Chem. – Eur. J. 2014, 20, 9918–9929. doi:10.1002/chem.201402623
Return to citation in text: [1] [2] -
Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954–6971. doi:10.1002/anie.200804497
Return to citation in text: [1] [2] [3] -
Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525–3550. doi:10.1039/c3cs60289c
Return to citation in text: [1] [2] [3] -
Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Hansen, J. A.; Becher, J.; Jeppesen, J. O.; Levillain, E.; Nielsen, M. B.; Petersen, B. M.; Petersen, J. C.; Şahin, Y. J. Mater. Chem. 2004, 14, 179–184. doi:10.1039/b310733g
Return to citation in text: [1] -
Wolfe, J. P.; Wagaw, S.; Marcoux, J.-F.; Buchwald, S. L. Acc. Chem. Res. 1998, 31, 805–818. doi:10.1021/ar9600650
Return to citation in text: [1] -
Hartwig, J. F. Angew. Chem., Int. Ed. 1998, 37, 2046–2067. doi:10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L
Return to citation in text: [1] -
Nygaard, S.; Hansen, C. N.; Jeppesen, J. O. J. Org. Chem. 2007, 72, 1617–1626. doi:10.1021/jo061962c
Return to citation in text: [1] -
D. Schlüter, K. R. Korsching, V. A. Azov, manuscript in preparation for Eur. J. Org. Chem.
Return to citation in text: [1] -
Lichtenberger, D. L.; Johnston, R. L.; Hinkelmann, K.; Suzuki, T.; Wudl, F. J. Am. Chem. Soc. 1990, 112, 3302–3307. doi:10.1021/ja00165a007
Return to citation in text: [1] -
Nielsen, M. B.; Jeppesen, J. O.; Lau, J.; Lomholt, C.; Damgaard, D.; Jacobsen, J. P.; Becher, J.; Stoddart, J. F. J. Org. Chem. 2001, 66, 3559–3563. doi:10.1021/jo010173m
Return to citation in text: [1] -
Connely, N. G.; Geiger, W. E. Chem. Rev. 1996, 96, 877–910. doi:10.1021/cr940053x
Return to citation in text: [1] -
Steiner, T. Angew. Chem., Int. Ed. 2002, 41, 48–76. doi:10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U
Return to citation in text: [1] -
Bryce, M. R.; Devonport, W.; Goldenberg, L. M.; Wang, C. Chem. Commun. 1998, 945–951. doi:10.1039/a800536b
Return to citation in text: [1] -
See Supporting Information for full details related to crystal structure refinement, ORTEP plots, and tables of close contacts.
Return to citation in text: [1] -
Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4945. doi:10.1021/cr030666m
Return to citation in text: [1]
| 32. | Bryce, M. R.; Devonport, W.; Goldenberg, L. M.; Wang, C. Chem. Commun. 1998, 945–951. doi:10.1039/a800536b |
| 33. | See Supporting Information for full details related to crystal structure refinement, ORTEP plots, and tables of close contacts. |
| 34. | Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4945. doi:10.1021/cr030666m |
| 1. | Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::AID-ANIE1372>3.0.CO;2-I |
| 2. | Yamada, J.; Sugimoto, T. TTF Chemistry. Fundamentals and Applications of Tetrathiafulvalene; Springer: Heidelberg, Germany, 2004. |
| 10. | Ballardini, R.; Balzani, V.; Becher, J.; Di Fabio, A.; Gandolfi, M. T.; Mattersteig, G.; Nielsen, M. B.; Raymo, F. M.; Rowan, S. J.; Stoddart, J. F.; White, A. J. P.; Williams, D. J. J. Org. Chem. 2000, 65, 4120–4126. doi:10.1021/jo0001941 |
| 20. | Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954–6971. doi:10.1002/anie.200804497 |
| 21. | Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525–3550. doi:10.1039/c3cs60289c |
| 7. | Li, Z.-T.; Stein, P. C.; Becher, J.; Jensen, D.; Mørk, P.; Svenstrup, N. Chem. – Eur. J. 1996, 2, 624–633. doi:10.1002/chem.19960020605 |
| 8. | Le Derf, F.; Mazari, M.; Mercier, N.; Levillain, E.; Trippé, G.; Riou, A.; Richomme, P.; Becher, J.; Garín, J.; Orduna, J.; Gallego-Planas, N.; Gorgues, A.; Sallé, M. Chem. – Eur. J. 2001, 7, 447–455. doi:10.1002/1521-3765(20010119)7:2<447::AID-CHEM447>3.0.CO;2-A |
| 9. | Azov, V. A.; Cordes, J.; Schlüter, D.; Dülcks, T.; Böckmann, M.; Doltsinis, N. L. J. Org. Chem. 2014, 79, 11714–11721. doi:10.1021/jo502469z |
| 24. | Wolfe, J. P.; Wagaw, S.; Marcoux, J.-F.; Buchwald, S. L. Acc. Chem. Res. 1998, 31, 805–818. doi:10.1021/ar9600650 |
| 25. | Hartwig, J. F. Angew. Chem., Int. Ed. 1998, 37, 2046–2067. doi:10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L |
| 5. | Fabre, J. M. Chem. Rev. 2004, 104, 5133–5150. doi:10.1021/cr0306440 |
| 6. | Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485 |
| 17. | Li, H.; Lambert, C. Chem. – Eur. J. 2006, 12, 1144–1155. doi:10.1002/chem.200500928 |
| 18. | Li, J.; Zhang, G.; Zhang, D.; Zheng, R.; Shi, Q.; Zhu, D. J. Org. Chem. 2010, 75, 5330–5333. doi:10.1021/jo1007306 |
| 19. | Solano, M. V.; DellaPia, E. A.; Jevric, M.; Schubert, C.; Wang, X.; van der Pol, C.; Kadziola, A.; Nørgaard, K.; Guldi, D. M.; Nielsen, M. B.; Jeppesen, J. O. Chem. – Eur. J. 2014, 20, 9918–9929. doi:10.1002/chem.201402623 |
| 3. | Becher, J.; Jeppesen, J. O.; Nielsen, K. Synth. Met. 2003, 133–134, 309–315. doi:10.1016/S0379-6779(02)00379-X |
| 4. | Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n |
| 20. | Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954–6971. doi:10.1002/anie.200804497 |
| 21. | Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525–3550. doi:10.1039/c3cs60289c |
| 17. | Li, H.; Lambert, C. Chem. – Eur. J. 2006, 12, 1144–1155. doi:10.1002/chem.200500928 |
| 18. | Li, J.; Zhang, G.; Zhang, D.; Zheng, R.; Shi, Q.; Zhu, D. J. Org. Chem. 2010, 75, 5330–5333. doi:10.1021/jo1007306 |
| 19. | Solano, M. V.; DellaPia, E. A.; Jevric, M.; Schubert, C.; Wang, X.; van der Pol, C.; Kadziola, A.; Nørgaard, K.; Guldi, D. M.; Nielsen, M. B.; Jeppesen, J. O. Chem. – Eur. J. 2014, 20, 9918–9929. doi:10.1002/chem.201402623 |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 14. | Yin, B.; Yang, Y.; Cong, Z.; Imafuku, K. Heterocycles 2004, 63, 1577–1584. doi:10.3987/COM-04-10083 |
| 15. | Balandier, J.-Y.; Chas, M.; Dron, P. I.; Goeb, S.; Canevet, D.; Belyasmine, A.; Allain, M.; Sallé, M. J. Org. Chem. 2010, 75, 1589–1599. doi:10.1021/jo902529e |
| 16. | Nygaard, S.; Leung, K. C.-F.; Aprahamian, I.; Ikeda, T.; Saha, S.; Laursen, B. W.; Kim, S.-Y.; Hansen, S. W.; Stein, P. C.; Flood, A. H.; Stoddart, J. F.; Jeppesen, J. O. J. Am. Chem. Soc. 2007, 129, 960–970. doi:10.1021/ja0663529 |
| 23. | Hansen, J. A.; Becher, J.; Jeppesen, J. O.; Levillain, E.; Nielsen, M. B.; Petersen, B. M.; Petersen, J. C.; Şahin, Y. J. Mater. Chem. 2004, 14, 179–184. doi:10.1039/b310733g |
| 12. | Jeppesen, J. O.; Takimiya, K.; Jensen, F.; Brimert, T.; Nielsen, K.; Thorup, N.; Becher, J. J. Org. Chem. 2000, 65, 5794–5805. doi:10.1021/jo000742a |
| 13. | Jeppesen, J. O.; Becher, J. Eur. J. Org. Chem. 2003, 3245–3266. doi:10.1002/ejoc.200300078 |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 11. | Souizi, A.; Robert, A.; Batail, P.; Ouahab, L. J. Org. Chem. 1987, 52, 1610–1611. doi:10.1021/jo00384a044 |
| 20. | Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954–6971. doi:10.1002/anie.200804497 |
| 21. | Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525–3550. doi:10.1039/c3cs60289c |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 27. | D. Schlüter, K. R. Korsching, V. A. Azov, manuscript in preparation for Eur. J. Org. Chem. |
| 12. | Jeppesen, J. O.; Takimiya, K.; Jensen, F.; Brimert, T.; Nielsen, K.; Thorup, N.; Becher, J. J. Org. Chem. 2000, 65, 5794–5805. doi:10.1021/jo000742a |
| 26. | Nygaard, S.; Hansen, C. N.; Jeppesen, J. O. J. Org. Chem. 2007, 72, 1617–1626. doi:10.1021/jo061962c |
| 12. | Jeppesen, J. O.; Takimiya, K.; Jensen, F.; Brimert, T.; Nielsen, K.; Thorup, N.; Becher, J. J. Org. Chem. 2000, 65, 5794–5805. doi:10.1021/jo000742a |
| 31. | Steiner, T. Angew. Chem., Int. Ed. 2002, 41, 48–76. doi:10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U |
| 30. | Connely, N. G.; Geiger, W. E. Chem. Rev. 1996, 96, 877–910. doi:10.1021/cr940053x |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 22. | Düker, M. H.; Schäfer, H.; Zeller, M.; Azov, V. A. J. Org. Chem. 2013, 78, 4905–4912. doi:10.1021/jo400502t |
| 1. | Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::AID-ANIE1372>3.0.CO;2-I |
| 28. | Lichtenberger, D. L.; Johnston, R. L.; Hinkelmann, K.; Suzuki, T.; Wudl, F. J. Am. Chem. Soc. 1990, 112, 3302–3307. doi:10.1021/ja00165a007 |
| 29. | Nielsen, M. B.; Jeppesen, J. O.; Lau, J.; Lomholt, C.; Damgaard, D.; Jacobsen, J. P.; Becher, J.; Stoddart, J. F. J. Org. Chem. 2001, 66, 3559–3563. doi:10.1021/jo010173m |
© 2015 Azov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)