Abstract
The design, development and application of an efficient procedure for the concise synthesis of the 1,3-syn- and anti-tetrahydropyrimidine cores of manzacidins are reported. The intramolecular allylic substitution reaction of a readily available joint urea-type substrate enables the facile preparation of both diastereomers in high yields. The practical application of this approach is demonstrated in the efficient and modular preparation of the authentic heterocyclic cores of manzacidins, structurally unique bromopyrrole alkaloids of marine origin. Additional features of this route include the stereoselective generation of the central amine core with an appending quaternary center by an asymmetric addition of a Grignard reagent to a chiral tert-butanesulfinyl ketimine following an optimized Ellman protocol and a cross-metathesis of a challenging homoallylic urea substrate, which proceeds in good yields in the presence of an organic phosphoric acid.

Graphical Abstract
Introduction
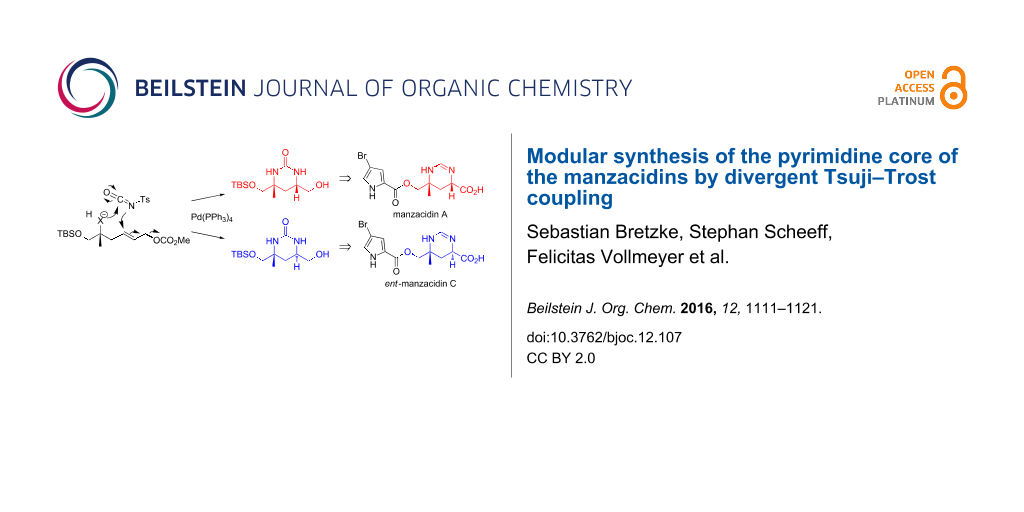
Chiral pyrimidine motifs constitute prevalent structural features in a variety of potent natural products and bioactive agents [1-5]. As exemplified by the marine natural products manzazidins A and C [2-5], they may be characterized by diverse configurations, including synthetically challenging quaternary centers. Owing to their pronounced biological activities, several synthetic routes have been reported to access these important substructures [6-22]. The manzacidins have first been isolated by the group of Kobayashi from the marine sponge Hymeniacidon sp. in the early nineties of the last century [2]. The compounds have demonstrated potent antifungal activity [3], and acted as α-adrenoceptor blockers, antagonists of the serotonergic receptor and/or actomyosin ATPase activators [23-25]. As shown in Figure 1 for the most prominent representatives, manzacidins A (1) and C (2), their unique architecture is characterized by an ester-linked bromopyrrole carboxylic acid and a tetrahydropyrimidine ring in which one of the amino groups is attached to a quaternary carbon center. Due to their intriguing structures in combination with the promising biological properties this class of bromopyrrole alkaloids has attracted great interest from synthetic chemists and a variety of elegant total syntheses has been reported [6-22]. Inspired by an innovative concept for heterocycles synthesis recently developed in our group [26-31], we became interested to devise a novel and a more versatile route to the central heterocyclic core of these marine metabolites. The method is based on a late-stage diversification strategy involving a Tsuji–Trost reaction of the urea-type joint precursor 5. In contrast to existing routes, this approach enables a more versatile elaboration of different configurations as present in the manzacidins and/or originally postulated for this class of marine natural products. Notably, the absolute configuration of manzacidin C was initially proposed as shown in Figure 1 [2] and subsequently revised by a total synthesis [6] which adds to the importance of a flexible route to such substructures. Herein we report in full detail the design, development and application of an innovative strategy for the high-yielding synthesis of 1,3-syn- and anti-configured tetrahydropyrimidinones, based on an allylic substitution reaction of a joint precursor 5. Subsequently this strategy is successfully applied to the synthesis of the authentic pyrimidine cores 3 and 4 of manzacidin A (1) and ent-manzacidin C (2).
![[1860-5397-12-107-1]](/bjoc/content/figures/1860-5397-12-107-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Modular concept for manzacidin synthesis based on a Tsuji–Trost coupling of joint intermediate 5.
Figure 1: Modular concept for manzacidin synthesis based on a Tsuji–Trost coupling of joint intermediate 5.
Results and Discussion
General synthetic concept
As part of our ongoing efforts to the design of novel tandem reactions for the synthesis of complex natural products [29,32-37], we have developed an innovative concept for heterocycles synthesis [26-31]. As shown in Scheme 1, this approach that further advances and generalizes several individual reports by other groups [38-43], is based on a sequential nucleophilic addition and an intramolecular allylic substitution reaction. It relies on the coupling of different homoallylic nucleophiles of general type 6 to diverse electrophiles 7 such as Michael acceptors, or heteroolefins as for example imines, carbonyls or allene homologs. The resulting homologated nucleophile 8 may then be trapped in an intramolecular fashion by a π-allyl complex, which may concomitantly form from 6 through activation of the homoallylic functionality with a suitable transition metal catalyst. According to this concept, variously substituted 6-membered heterocycles of type 9 may be obtained in a general and concise fashion. Notably, this anionic relay process may directly generate up to four new stereogenic centers and thus demonstrates a high increase in structural complexity from readily available starting materials.
![[1860-5397-12-107-i1]](/bjoc/content/inline/1860-5397-12-107-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General concept for heterocycles synthesis based on a nucleophilic addition and Tsuji–Trost coupling.
Scheme 1: General concept for heterocycles synthesis based on a nucleophilic addition and Tsuji–Trost couplin...
Evaluation of the concept by a model study
As a prelude to the targeted substitution pattern of the manzacidins, we first evaluated the applicability of this process for a modular synthesis of 1,3-syn- and anti-tetrahydropyrimidinones using the simplified amine substrate 12. Parts of this model study have already been reported in preliminary form [31]. Homoallylic amines of type 12 may be efficiently obtained through multicomponent reactions. These involve the nucleophilic allylation of imines which may be generated in situ by the condensation of an amine and a carbonyl compound. As shown in Scheme 2, two such procedures were evaluated within the preliminary study. The first protocol that we analyzed was reported by the group of Tian. It involves a four-component coupling of aldehyde 10 with CbzCl for activation of the nitrogen source, HMDS and allyltrimethylsilane (11) in the presence of catalytic amounts of FeSO4 [44]. In our hands, this process enabled an efficient access to the desired homoallylic amine 12 in essentially quantitative yields. The other protocol was reported by Phukan and involves an iodine-catalyzed condensation of aldehyde 10 with benzylcarbamate and allyltrimethylsilane (11) [45]. Unfortunately, this route was found to be less effective in terms of isolated yields and scalability. Thus, the iron-catalyzed procedure was applied and multigram quantities of 12 were readily obtained.
![[1860-5397-12-107-i2]](/bjoc/content/inline/1860-5397-12-107-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of homoallylic alcohol 12 by multi-component reactions.
Scheme 2: Synthesis of homoallylic alcohol 12 by multi-component reactions.
As shown in Scheme 3, we next focused on the further derivatization of amine 12 towards suitably functionalized urea substrates 15 or 19. Inspired by a work of Garcia [39], we initially intended to use isocyanate for both, the introduction of the urea motif and for the functionalization of the terminal homoallylic alcohol. Consequently, we evaluated the conversion of 14 to 15. The required substrate 14 was prepared from amine 12 by cross-metathesis with 2-butene-1,4-diol (13) in the presence of Grubbs-II catalyst 21. However, in the subsequent coupling reactions of 14 with TsNCO it became apparent that this homoallylic amine was too unreactive to enable a double addition to access 15 directly. Therefore, a stepwise approach towards 19 was pursued instead. This involved either a coupling of 12 first with isocyanate to give 16 followed by a cross-metathesis or starting with the cross-metathesis to 18 and subsequent installment of the urea motif. As shown in the table inserted in Scheme 3 for selected cross-metatheses of Cbz-protected amide 12 and its urea-derivative 16 with butene 17, a different reactivity of 12 and 16 was observed. While 16 proved too unreactive for the coupling reaction under various conditions (e.g., entries 1 and 2), the homologation of the Cbz-protected amine 12 to 18 could be realized. Preparative useful yields (69%) were obtained with Grubbs-II catalyst (21) in toluene at elevated temperatures (entry 3), while lower conversions were observed with other catalysts (20, 22) or in dichloromethane (entries 4 and 5). Finally, for the installment of the required urea motif into 18, tosylisocyanate in combination with strong bases was required to achieve useful degrees of conversion towards the desired precursor 19. The best results were obtained with BuLi, as previously communicated [31], while weaker bases (NEt3, LHMDS, DBU, proton sponge) and less electron-deficient isocyanates resulted in lower yields.
![[1860-5397-12-107-i3]](/bjoc/content/inline/1860-5397-12-107-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of urea-type cyclization precursor 19.
Scheme 3: Preparation of urea-type cyclization precursor 19.
We then turned our attention to the pivotal intramolecular allylic substitution reaction of 19 to access syn- and anti-pyrimidinones 23 and 24. As previously reported [31], this diastereodivergent coupling could indeed be realized as shown in Scheme 4. Based on a report of Garcia for a related system we first evaluated Pd2(dba)3 with different phosphite ligands [39]. However, the best results were obtained with the stable catalyst Pd(PPh3)4 and depending on the solvent used, either the syn-isomer 23 or the anti-isomer 24 could be selectively obtained.
![[1860-5397-12-107-i4]](/bjoc/content/inline/1860-5397-12-107-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Stereodivergent synthesis of 1,3-syn- and anti-tetrahydropyrimidinones [31].
Scheme 4: Stereodivergent synthesis of 1,3-syn- and anti-tetrahydropyrimidinones [31].
Application of the concept for manzacidin core synthesis
After proofing the general adaptability of our synthetic concept, we next evaluated the applicability of this procedure for the synthesis of the authentic manzacidin substrate. As shown in Scheme 5, we first focused on the stereoselective synthesis of the chiral amine core of these alkaloids. For the synthesis of the nitrogen appending the quaternary center we tested a method developed by the Ellman group [46,47], which relies on an asymmetric addition of organometallic reagents to enantiopure tert-butanesulfinyl ketimines of type 29 and 30. Although the group of Lee had already communicated the synthesis of 33 using this approach [48], no full details have been given. In addition, the reported yields were only moderate and the stereochemistry appeared not to have been rigorously assigned. Therefore, we evaluated this type of asymmetric addition in more general terms and analyzed the addition reactions of allylmagnesium bromide both to 29 and 30. Notably, this route would allow to access all possible stereoisomers of the manzacidins, in agreement with the stereochemical diversity of this class of natural products. In detail, the synthesis of 29 and 30 involved a condensation of hydroxyacetone (25)-derived ketone 26 [49] with SS- and RS-tert-butanesulfinamides 27 and 28, respectively. As an improvement to the original procedure [46-48], we applied Ti(OiPr)4 as Lewis acid instead of the reported Ti(OEt)4, which resulted in higher yields and a more reliable process in our hands. In agreement with the results of Lee the addition of allylmagnesium bromide to 30 lead to 33 in only moderate yields and low selectivity towards 34. We then studied the coupling of 29 in more detail to target amine 31 that bears the correct configuration required for manzacidin A. Possibly, the higher selectivity observed for the conversion of 29 as compared to 30 may be due to initial problems during the work-up. Finally, the addition could be effected giving the desired diastereomer 31 in high yields (72%) and the minor isomer 32 that was likewise obtained (24%) could be readily removed by column chromatography. The configuration of 31 was initially assigned by Mosher ester analysis of the free amine 36 (Scheme 6) and finally proven in an indirect manner by an X-ray crystallography of the minor diastereomer 32. Within the course of this study also an X-ray structure of tert-butylsulfinylamine 28 was obtained. Remarkably, these types of substances have not been broadly evaluated by X-ray structural analysis which adds to the importance of this general evaluation.
![[1860-5397-12-107-i5]](/bjoc/content/inline/1860-5397-12-107-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Stereoselective synthesis of all possible stereoisomers of the manzacidin core amine by asymmetric addition to chiral tert-butanesulfinyl ketimines.
Scheme 5: Stereoselective synthesis of all possible stereoisomers of the manzacidin core amine by asymmetric ...
Next, we focused on further homologation towards a suitably functionalized urea precursor 5 for the envisioned Tsuji–Trost cyclization. As shown in Scheme 6, this involved an acidic cleavage of the sulfinamide followed by basic treatment to give free amine 35. After protection of the primary hydroxy group as TBS ether, we first evaluated the synthesis of derivative 40, in analogy to our model study. Accordingly, the free amine 36 was Cbz-protected following the Schotten–Baumann method [50]. The obtained amide 37 was then homologated by cross-metathesis with butenedicarboxylate 17 in the presence of Grubbs-II catalyst (21) applying our conditions developed above (Scheme 3). However, with the resulting homologated amide 38 in hand we were not able to install the required urea moiety with tosylisocyanate, despite considerable efforts with various bases, solvents or variation of temperature and equivalents. These results again demonstrated the difficulties to install the urea function in a sterically hindered and electronically unreactive Cbz-protected amine substrate, which is in agreement with our observations above. Therefore, we decided to continue our route with the free amine 36 instead, which was directly coupled with TSNCO to give 39 in high yield. The reaction took place even without an additional base, which shows the strong influence of the amine protective group on this type of condensation. Importantly, at this stage, the structure of 39 was fully confirmed by X-ray crystallography. As shown in Figure 2, this urea derivative is present as an unsymmetrical dimer in the crystal lattice, which is stabilized by two hydrogen bonds between the urea oxygen atoms and the tosyl-protected nitrogens. This also unambiguously confirms the absolute configuration of 39 and corroborates our prediction of the asymmetric adduct 31.
![[1860-5397-12-107-i6]](/bjoc/content/inline/1860-5397-12-107-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of the authentic cyclization precursor 5.
Scheme 6: Synthesis of the authentic cyclization precursor 5.
![[1860-5397-12-107-2]](/bjoc/content/figures/1860-5397-12-107-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
We then attempted the installation of the required allylic carbonate on 39 by cross-metathesis with 17. However, initial attempts following our protocol developed above with Grubbs-II catalyst (21) resulted in only moderate conversion (inserted table in Scheme 6, entry 1). Also, the application of other catalysts with or without additional additives to impede a possibly unfavorable amine coordination of the reactive ruthenium intermediates [51] did not improve the reaction outcome (entries 2–4). Following reports from Nolan and Prunet [52], as well as from Steinke and Vilar [53] we finally evaluated tricyclohexylphosphane oxides and organic phosphoric acid, which had been reported to have beneficial effects in the cross-metathesis of related substrates. In the presence of catalytic amounts of phosphoric acid 41 [53], the coupling of 39 with 17 could indeed be realized in useful yields in a reliable fashion. Optimal results included treatment of 39 with 2.5 equiv of dicarbonate 17, 50 mol % naphthylphosphoric acid and 10 mol % Grubbs-II catalyst, giving the desired urea derivative 5 in good yield (67%), considering the general difficulties observed for such substrates in cross-metathesis reactions.
With precursor 5 in hand the desired cyclization towards 42 and 43 could then be efficiently realized in a straightforward manner giving the desired syn- and anti- tetrahydropyrimidinones in a joint fashion with a ratio of 1.5:1. Following the protocol developed above, excellent yields (94%) were obtained in this coupling. As compared to the model substrate 19 (see Scheme 4) no selectivity was observed in this coupling, which could also not be modified by other solvents. Possibly this may be due to the missing Cbz group of 5 as compared to 19. The configuration of both products was assigned by NMR methods based on characteristic NOE correlations and vicinal coupling constants as shown in Scheme 7. For further conversion to key intermediates 3 and 4, the tosyl groups of 42 and 43 were removed with SmI2 [54,55] giving the free amides 44 and 45. The terminal double bonds were then oxidized by dihydroxylation with OsO4 and periodate cleavage [56,57], and the resulting aldehydes (not shown) were reduced to the terminal alcohols with NaBH4, giving the desired pyrimidinones 3 and 4. These compounds represent key intermediates which may be transformed into the targeted natural products 1 and 2 following previously established protocols [6,31].
![[1860-5397-12-107-i7]](/bjoc/content/inline/1860-5397-12-107-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Divergent Tsuji–Trost coupling and completion of the synthesis of authentic pyrimidinones 3 and 4.
Scheme 7: Divergent Tsuji–Trost coupling and completion of the synthesis of authentic pyrimidinones 3 and 4.
Conclusion
In summary, we have reported in full details the design, development and application of an efficient method for the synthesis of the tetrahydropyrimidinone core of the manzacidins by a divergent intramolecular allylic substitution reaction. The application of this approach enabled a highly concise access to the authentic heterocyclic cores of the manzacidins, structurally unique natural products of marine origin. Additional notable features of our modular route also include the generation of an amine appending quaternary center by an optimized Ellman protocol for the asymmetric allyl-Grignard addition to enantiopure tert-butanesulfinyl ketimines and an efficient cross-metathesis of an unreactive urea substrate in the presence of an organic phosphoric acid. It is expected that these strategies and tactics will find applications in functional target synthesis and stimulate further studies for modular heterocycle synthesis.
Experimental
Preparation of 31 by asymmetric addition of allylmagnesium bromide to 29
In a flame-dried flask (SS)-N-(1-((tert-butyldimethylsilyl)oxy)propan-2-ylidene)-2-methylpropane-2-sulfinamide (29, 1.37 g, 4.70 mmol, 1.0 equiv) was dissolved in 12 mL toluene and the solution was cooled to −78 °C. To this mixture allylmagnesium bromide (1.0 M in Et2O, 7.05 mL, 7.05 mmol, 1.5 equiv) was slowly added and the reaction was stirred for 1 h at −78 °C. The reaction was quenched with a solution of saturated Na2SO4, warmed to rt, filtered, washed with ethyl acetate and finally purified by column chromatography on silica gel (120 g) with ethyl acetate/hexane 1:9 as eluent, which yielded the desired diasteromers 31 (major diastereomer) and 32 (minor diastereomer) as light yellow oils (96 %, dr = 1:3). Major diastereomer SSR (1.13 g, 3.39 mmol, 72%): Rf 0.1 (ethyl acetate/hexane 1:9); [α]20D +58.2 (c 1.00, CHCl3); 1H NMR (300.13 MHz, CDCl3) δ 0.06 (s, 6H), 0.91 (s, 9H), 1.19 (s, 12H), 2.48 (dd, J = 4.1 Hz, 7.4 Hz, 2H), 3.32 (d, J = 9.3 Hz, 1H), 3.49 (d, J = 9.3 Hz, 1H), 3.72 (bs, 1H), 5.11 (d, J = 10.4 Hz, 1H), 5.12 (d, J = 16.7 Hz, 1H), 5.80 (ddt, J = 7.4 Hz, 10.4 Hz, 17.8 Hz, 1H); 13C NMR (75.47 MHz, CDCl3) δ −5.5, 18.2, 22.1, 22.6, 25.8, 43.0, 55.5, 58.1, 69.2, 118.7, 133.8; HRMS–FAB (m/z): [M + H]+ calcd for C16H36NO2SSi, 334.2231; found, 334.2227. Minor diastereomer SSS (371 mg, 1.11 mmol, 24%): Rf 0.13 (ethyl acetate/hexane 1:9); [α]20D +39.2 (c 1.00, CHCl3); 1H NMR (300.13 MHz, CDCl3) δ 0.07 (s, 3H), 0.08 (s, 3H), 0.91 (s, 9H), 1.20 (s, 9H), 1.28 (s, 3H), 2.22 (dd, J = 8.0 Hz, 13.7 Hz, 1H), 2.37 (dd, J = 6.7 Hz, 14.0 Hz, 1H), 3.48 (d, J = 9.3 Hz, 1H), 3.53 (d, J = 9.6 Hz, 1H), 3.77 (bs, 1H), 5.09 (d, J = 17.8 Hz, 1H), 5.10 (d, J = 10.7 Hz, 1H), 5.78 (ddt, J = 8.0 Hz, 10.7 Hz, 17.3 Hz, 1H); 13C NMR (75.47 MHz, CDCl3) δ −5.6, 18.2, 22.3, 22.7, 25.8, 43.1, 55.6, 58.1, 69.9, 118.5, 133.6; HRMS–ESI (m/z): [M + H]+ calcd for C16H36NO2SSi, 334.2231; found, 334.2231.
Preparation of 39 by addition of TsNCO to amine 36
p-TsNCO (0.6 mL, 4.13 mmol, 1.1 equiv) was slowly added to a stirred solution of (R)-1-((tert-butyldimethylsilyl)oxy)-2-methylpent-4-en-2-amine (36, 902 mg, 3.93 mmol) in dry THF (3.9 mL) at 0 °C and stirring was continued at rt for 5 h. The solvent was removed under reduced pressure and purification of the residue by column chromatography on silica gel (cyclohexane/ethyl acetate 4:1) yielded the desired product (1.61 g, 3.77 mmol, 96%) as a colorless solid. Rf 0.29 (cyclohexane/ethyl acetate 4:1); mp 84 °C; [α]20D −2.3 (c 0.5, CHCl3); 1H NMR (300.13 MHz, CDCl3) δ 0.09 (s, 6H), 0.93 (s, 9H), 1.24 (s, 3H), 2.43 (d, J = 8.0 Hz, 2H), 2.44 (s, 3H), 3.44 (d, J = 9.8 Hz, 1H), 3.58 (d, J = 9.8 Hz, 1H), 4.99 (dd, J = 10.1, 2.1 Hz, 1H), 5.04 (dd, J = 17.2, 2.1 Hz, 1H), 5.58 (ddt, J = 17.2, 10.1, 8.0 Hz, 1H), 6.84 (brs, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H); 13C NMR (75.47 MHz, CDCl3) δ −5.4, 18.5, 21.4, 21.8, 26.0, 40.1, 57.3, 67.8, 118.8, 127.2, 129.9, 133.3, 137.0, 144.7, 150.3; HRMS–ESI (m/z): [M + Na]+ calcd for C20H34N2NaO4SSi, 449.1901; found, 449.1892. CCDC 1461909 (39) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Preparation of 5 by cross-metathesis of 39 with 17
To a solution of (R)-N-((1-((tert-butyldimethylsilyl)oxy)-2-methylpent-4-en-2-yl)carbamoyl)-4-methylbenzenesulfonamide (39, 50.0 mg, 0.12 mmol, 1.0 equiv), (Z)-(but-2-ene-1,4-diyl)dimethyl dicarbonate (17, 60.0 mg, 0.29 mmol, 2.5 equiv) and naphthylphosphoric acid (41, 15.0 mg, 67.0 µmol, 0.5 equiv) in dry and degassed dichloromethane (1 mL) was added Grubbs-II catalyst (21, 10.0 mg, 11.8 µmol, 10 mol %) and the resulting mixture was stirred overnight at 50 °C under an argon atmosphere. Concentration in vacuo and purification by column chromatography on silica gel (10 g) with ethyl acetate/hexane 1:9 as eluent yielded the desired allylic carbonate as brown oil (41.3 mg, 80.4 µmol, 67%): Rf 0.30 (ethyl acetate/hexane 1:3); [α]20D −2.9 (c 1.00, CHCl3); 1H NMR (300.13 MHz, CDCl3) δ 0.09 (s, 6H), 0.93 (s, 9H), 1.24 (s, 3H), 2.44 (m, 5H), 3.44 (d, J = 9.8 Hz, 1H), 3.56 (d, J = 9.8 Hz, 1H), 3.79 (s, 3H), 4.49 (d, J = 4.7 Hz, 2H), 5.60 (m, 2H), 6.87 (s, 1H), 7.31 (d, J = 8.1 Hz, 2H), 7.77 (d, J = 8.3 Hz, 2H), 8.42 (bs, 1H); 13C NMR (75.47 MHz, CDCl3) δ −5.6, 18.3, 21.3, 21.6, 25.8, 38.5, 54.8, 57.1, 67.7, 68.2, 127.0, 127.5, 129.9, 131.0, 136.7, 144.8, 149.8, 155.6; HRMS–ESI (m/z): [M + H]+ calcd for C23H39N2O7SSi, 515.2242; found, 515.2247; HRMS–ESI (m/z): [M + Na]+ calcd for C23H38N2O7SSiNa, 537.2061; found, 537.2065.
Tsuji–Trost coupling of 5 to 42 and 43
A solution of Pd(PPh3)4 (432 mg, 3.73 µmol, 20 mol %) in dry THF (300 mL) was added to a stirred solution of (R,E)-6-((tert-butyldimethylsilyl)oxy)-5-methyl-5-(3-tosylureido)hex-2-en-1-ylmethyl carbonate (5, 959 mg, 1.86 mmol) in dry THF (300 mL) at rt and stirring was continued for 18 h until the color of the solution changed from yellow to red. The solvent was removed under reduced pressure and purification of the residue by column chromatography on silica gel (cyclohexane/ethyl acetate 4:1) yielded the desired products 42 and 43 (766 mg, 1.75 mmol, 94%, dr 1:1.5 anti/syn) as off-white solids. 42: mp 129 °C; [α]20D −20.3 (c 0.5, CHCl3); 1H NMR (400.13 MHz, CDCl3) δ 0.00 (s, 6H), 0.86 (s, 9H), 1.18 (s, 3H), 1.92 (dd, J = 14.2, 6.3 Hz, 1H), 2.10 (dd, J = 14.2, 3.6 Hz, 1H), 2.39 (s, 3H), 3.35–3.43 (m, 2H), 5.12–5.16 (m, 1H), 5.18 (dd, J = 10.5, 1.5 Hz, 1H), 5.25 (dd, J = 17.2, 1.5 Hz, 1H), 5.45 (brs, 1H), 5.79 (ddd, J = 17.2, 10.5, 5.4 Hz, 1H), 7.25 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H); 13C NMR (100.62 MHz, CDCl3) δ −5.4, −5.4, 18.2, 21.7, 25.9, 27.1, 35.8, 55.1, 56.3, 69.1, 116.8, 129.0, 129.1, 137.1, 137.2, 144.2, 151.4; HRMS–EI (m/z): [M – C4H9]+ calcd for C17H25N2NaO7SSi, 381.1304; found, 381.1307. 43: mp 127 °C; [α]20D −12.8 (c 0.5, CHCl3); 1H NMR (400.13 MHz, CDCl3) δ 0.02 (s, 3H), 0.03 (s, 3H), 0.85 (s, 9H), 1.20 (s, 3H), 1.89 (dd, J = 13.9, 3.2 Hz, 1H), 2.05 (dd, J = 13.9, 5.8 Hz, 1H), 2.05 (dd, J = 13.9, 5.8 Hz, 1H), 2.39 (s, 3H), 3.25 (d, J = 9.4 Hz, 1H), 3.34 (d, J = 9.4 Hz, 1H), 5.30–5.17 (m, 4H), 5.88 (ddd, J = 16.9, 10.5, 6.0 Hz, 1H), 7.25 (d, J = 8.3 Hz, 2 H), 7.90 (d, J = 8.3 Hz, 2H); 13C NMR (100.62 MHz, CDCl3) δ −5.4, −5.5, 18.3, 21.7, 25.6, 25.9, 36.3, 55.0, 56.3, 71.6, 117.0, 129.0, 129.2, 137.1, 144.2, 151.3; HRMS–EI (m/z): [M – C4H9]+ calcd for C17H25N2NaO7SSi, 381.1307; found, 381.1309.
Supporting Information
| Supporting Information File 1: Full experimental details, characterization data of all products, copies of 1H and 13C NMR spectra and X-ray crystallographic data for 28, 32 and 39. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Patil, A. D.; Kumar, N. V.; Kokke, W. C.; Bean, M. F.; Freyer, A. J.; De Brosse, C.; Mai, S.; Truneh, A.; Faulkner, D. J.; Carte, B.; Breen, A. L.; Hertzberg, R. P.; Johnson, P. K.; Westley, J. W.; Potts, B. C. M. J. Org. Chem. 1995, 60, 1182–1188. doi:10.1021/jo00110a021
Return to citation in text: [1] -
Kobayashi, J.; Kanda, F.; Ishibashi, M.; Shigemori, H. J. Org. Chem. 1991, 56, 4574–4576. doi:10.1021/jo00014a052
Return to citation in text: [1] [2] [3] [4] -
Tsukamoto, S.; Tane, K.; Ohta, T.; Matsunaga, S.; Fusetani, N.; van Soest, R. W. M. J. Nat. Prod. 2001, 64, 1576–1578. doi:10.1021/np010280b
Return to citation in text: [1] [2] [3] -
Jahn, T.; König, G. M.; Wright, A. D.; Wörheide, G.; Reitner, J. Tetrahedron Lett. 1997, 38, 3883–3884. doi:10.1016/S0040-4039(97)00846-0
Return to citation in text: [1] [2] -
Aiello, A.; D’Esposito, M.; Fattourusso, E.; Menna, M.; Müller, W. E. G.; Perović-Ottstadt, S.; Schröder, H. C. Bioorg. Med. Chem. 2006, 14, 17–24. doi:10.1016/j.bmc.2005.07.057
Return to citation in text: [1] [2] -
Namba, K.; Shinada, T.; Teramoto, T.; Ohfune, Y. J. Am. Chem. Soc. 2000, 122, 10708–10709. doi:10.1021/ja002556s
Return to citation in text: [1] [2] [3] [4] -
Wehn, P. M.; Du Bois, J. J. Am. Chem. Soc. 2002, 124, 12950–12951. doi:10.1021/ja028139s
Return to citation in text: [1] [2] -
Woo, J. C. S.; MacKay, D. B. Tetrahedron Lett. 2003, 44, 2881–2883. doi:10.1016/S0040-4039(03)00431-3
Return to citation in text: [1] [2] -
Drouin, C.; Woo, J. C. S.; MacKay, D. B.; Lavigne, R. M. A. Tetrahedron Lett. 2004, 45, 7197–7199. doi:10.1016/j.tetlet.2004.08.038
Return to citation in text: [1] [2] -
Kano, T.; Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 2174–2175. doi:10.1021/ja056851u
Return to citation in text: [1] [2] -
Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2006, 128, 3928–3930. doi:10.1021/ja060312n
Return to citation in text: [1] [2] -
Sibi, M. P.; Stanley, L. M.; Soeta, T. Org. Lett. 2007, 9, 1553–1556. doi:10.1021/ol070364x
Return to citation in text: [1] [2] -
Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2007, 9, 1765–1767. doi:10.1021/ol0704789
Return to citation in text: [1] [2] -
Tran, K.; Lombardi, P. J.; Leighton, J. L. Org. Lett. 2008, 10, 3165–3167. doi:10.1021/ol8011869
Return to citation in text: [1] [2] -
Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2010, 12, 2170. doi:10.1021/ol1006339
Return to citation in text: [1] [2] -
Ichikawa, Y.; Okumura, K.; Matsuda, Y.; Hasegawa, T.; Nakamura, M.; Fujimoto, A.; Masuda, T.; Nakano, K.; Kotsuki, H. Org. Biomol. Chem. 2012, 10, 614–622. doi:10.1039/C1OB06559A
Return to citation in text: [1] [2] -
Sankar, K.; Rahman, H.; Das, P. P.; Bhimireddy, E.; Sridhar, B.; Mohapatra, D. K. Org. Lett. 2012, 14, 1082–1085. doi:10.1021/ol203466m
Return to citation in text: [1] [2] -
Shinada, T.; Oe, K.; Ohfune, Y. Tetrahedron Lett. 2012, 53, 3250–3253. doi:10.1016/j.tetlet.2012.04.042
Return to citation in text: [1] [2] -
Yoshimura, T.; Kinoshita, T.; Yoshioka, H.; Kawabata, T. Org. Lett. 2013, 15, 864–867. doi:10.1021/ol303568f
Return to citation in text: [1] [2] -
Nagatomo, M.; Nishiyama, H.; Fujino, H.; Inoue, M. Angew. Chem., Int. Ed. 2015, 127, 1557–1561. doi:10.1002/ange.201410186
Return to citation in text: [1] [2] -
Hashimoto, T.; Maruoka, K. Org. Biomol. Chem. 2008, 6, 829–835. doi:10.1039/B716062C
Return to citation in text: [1] [2] -
Ohfune, Y.; Oe, K.; Namba, K.; Shinada, T. Heterocycles 2012, 85, 2617–2649. doi:10.3987/REV-12-746
Return to citation in text: [1] [2] -
Urban, S.; de Almeida Leone, P.; Carroll, A. R.; Fechner, G. A.; Smith, J.; Hooper, J. N. A.; Quinn, R. J. J. Org. Chem. 1999, 64, 731–735. doi:10.1021/jo981034g
Return to citation in text: [1] -
Kobayashi, J.; Inaba, K.; Tsuda, M. Tetrahedron 1997, 53, 16679–16682. doi:10.1016/S0040-4020(97)10097-7
Return to citation in text: [1] -
Kato, T.; Shizuri, Y.; Izumida, H.; Yokoyama, A.; Endo, M. Tetrahedron Lett. 1995, 36, 2133–2136. doi:10.1016/0040-4039(95)00194-H
Return to citation in text: [1] -
Herkommer, D.; Schmalzbauer, B.; Menche, D. Nat. Prod. Rep. 2014, 31, 456–467. doi:10.1039/C3NP70093C
Return to citation in text: [1] [2] -
Tang, B.; Wang, L.; Menche, D. Synlett 2013, 24, 625–629. doi:10.1055/s-0032-1318300
Return to citation in text: [1] [2] -
Wang, L.; Menche, D. J. Org. Chem. 2012, 77, 10811–10823. doi:10.1021/jo302102x
Return to citation in text: [1] [2] -
Wang, L.; Menche, D. Angew. Chem. 2012, 124, 9559–9562. doi:10.1002/ange.201203911
Angew. Chem., Int. Ed. 2012, 51, 9425–9427. doi:10.1002/anie.201203911
Return to citation in text: [1] [2] [3] -
Wang, L.; Li, P.; Menche, D. Angew. Chem. 2010, 122, 9456–9460. doi:10.1002/ange.201003304
Angew. Chem., Int. Ed. 2010, 49, 9270–9273. doi:10.1002/anie.201003304
Return to citation in text: [1] [2] -
Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Debnar, T.; Dreisigacker, S.; Menche, D. Chem. Commun. 2013, 49, 725–727. doi:10.1039/C2CC37678D
Return to citation in text: [1] -
Dieckmann, M.; Menche, D. Org. Lett. 2013, 15, 228–231. doi:10.1021/ol3033303
Return to citation in text: [1] -
Schrempp, M.; Thiede, S.; Herkommer, D.; Gansäuer, A.; Menche, D. Chem. – Eur. J. 2015, 21, 16266–16271. doi:10.1002/chem.201502263
Return to citation in text: [1] -
Thiede, S.; Winterscheid, P. M.; Hartmann, J.; Schnakenburg, G.; Essig, S.; Menche, D. Synthesis 2016, 48, 697–709. doi:10.1055/s-0035-1561278
Return to citation in text: [1] -
Herkommer, D.; Thiede, S.; Wosniok, P. R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. J. Am. Chem. Soc. 2015, 137, 4086–4089. doi:10.1021/jacs.5b01894
Return to citation in text: [1] -
Menche, D.; Arikan, F.; Li, J.; Rudolph, S. Org. Lett. 2007, 9, 267–270. doi:10.1021/ol062715y
Return to citation in text: [1] -
Alouane, N.; Boutier, A.; Baron, C.; Vrancken, E.; Mangeney, P. Synthesis 2006, 885–889. doi:10.1055/s-2006-926340
Return to citation in text: [1] -
Broustal, G.; Ariza, X.; Campagne, J.-M.; Garcia, J.; Georges, Y.; Marinetti, A.; Robiette, R. Eur. J. Org. Chem. 2007, 4293–4297. doi:10.1002/ejoc.200700503
Return to citation in text: [1] [2] [3] -
Fraunhoffer, K. J.; White, M. C. J. Am. Chem. Soc. 2007, 129, 7274–7276. doi:10.1021/ja071905g
Return to citation in text: [1] -
Amador, M.; Ariza, X.; Garcia, J.; Sevilla, S. Org. Lett. 2002, 4, 4511–4514. doi:10.1021/ol0270428
Return to citation in text: [1] -
Butler, D. C. D.; Inman, G. A.; Alper, H. J. Org. Chem. 2000, 65, 5887–5890. doi:10.1021/jo000608q
Return to citation in text: [1] -
Xie, Y.; Yu, K.; Gu, Z. J. Org. Chem. 2014, 79, 1289–1302. doi:10.1021/jo402681z
Return to citation in text: [1] -
Song, Q.-Y.; Yang, B.-L.; Tian, S.-K. J. Org. Chem. 2007, 72, 5407–5410. doi:10.1021/jo0704558
Return to citation in text: [1] -
Phukan, P. J. Org. Chem. 2004, 69, 4005–4006. doi:10.1021/jo0498462
Return to citation in text: [1] -
Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2
Return to citation in text: [1] [2] -
Tang, T. P.; Volkman, S. K.; Ellman, J. A. J. Org. Chem. 2001, 66, 8772–8778. doi:10.1021/jo0156868
Return to citation in text: [1] [2] -
Yendapally, R.; Lee, R. E. Bioorg. Med. Chem. Lett. 2008, 18, 1607–1611. doi:10.1016/j.bmcl.2008.01.065
Return to citation in text: [1] [2] -
Li, H.; Hong, J.-H. Bull. Korean Chem. Soc. 2008, 29, 847–850. doi:10.5012/bkcs.2008.29.4.847
Return to citation in text: [1] -
Herrington, P. M.; Owen, C. E.; Gage, J. R. Org. Process Res. Dev. 2001, 5, 80–83. doi:10.1021/op0002949
Return to citation in text: [1] -
Formentín, P.; Gimeno, N.; Steinke, J. H. G.; Vilar, R. J. Org. Chem. 2005, 70, 8235–8238. doi:10.1021/jo051120y
Return to citation in text: [1] -
Bourgeois, D.; Pancrazi, A.; Nolan, S. P.; Prunet, J. J. Organomet. Chem. 2002, 643–644, 247–252. doi:10.1016/S0022-328X(01)01269-4
Return to citation in text: [1] -
Gimeno, N.; Formentín, P.; Steinke, J. H. G.; Vilar, R. Eur. J. Org. Chem. 2007, 918–924. doi:10.1002/ejoc.200600908
Return to citation in text: [1] [2] -
Goulaouic-Dubois, C.; Guggisberg, A.; Hesse, M. Tetrahedron 1995, 51, 12573–12582. doi:10.1016/0040-4020(95)00811-L
Return to citation in text: [1] -
Knowles, H. S.; Parsons, A. F.; Pettifer, R. M.; Rickling, S. Tetrahedron 2000, 56, 979–988. doi:10.1016/S0040-4020(00)00025-9
Return to citation in text: [1] -
Pappo, R.; Allen, D. S., Jr.; Lemieux, R. U.; Johnson, W. S. J. Org. Chem. 1956, 21, 478–479. doi:10.1021/jo01110a606
Return to citation in text: [1] -
Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett. 2004, 6, 3217–3219. doi:10.1021/ol0400342
Return to citation in text: [1]
| 52. | Bourgeois, D.; Pancrazi, A.; Nolan, S. P.; Prunet, J. J. Organomet. Chem. 2002, 643–644, 247–252. doi:10.1016/S0022-328X(01)01269-4 |
| 53. | Gimeno, N.; Formentín, P.; Steinke, J. H. G.; Vilar, R. Eur. J. Org. Chem. 2007, 918–924. doi:10.1002/ejoc.200600908 |
| 53. | Gimeno, N.; Formentín, P.; Steinke, J. H. G.; Vilar, R. Eur. J. Org. Chem. 2007, 918–924. doi:10.1002/ejoc.200600908 |
| 1. | Patil, A. D.; Kumar, N. V.; Kokke, W. C.; Bean, M. F.; Freyer, A. J.; De Brosse, C.; Mai, S.; Truneh, A.; Faulkner, D. J.; Carte, B.; Breen, A. L.; Hertzberg, R. P.; Johnson, P. K.; Westley, J. W.; Potts, B. C. M. J. Org. Chem. 1995, 60, 1182–1188. doi:10.1021/jo00110a021 |
| 2. | Kobayashi, J.; Kanda, F.; Ishibashi, M.; Shigemori, H. J. Org. Chem. 1991, 56, 4574–4576. doi:10.1021/jo00014a052 |
| 3. | Tsukamoto, S.; Tane, K.; Ohta, T.; Matsunaga, S.; Fusetani, N.; van Soest, R. W. M. J. Nat. Prod. 2001, 64, 1576–1578. doi:10.1021/np010280b |
| 4. | Jahn, T.; König, G. M.; Wright, A. D.; Wörheide, G.; Reitner, J. Tetrahedron Lett. 1997, 38, 3883–3884. doi:10.1016/S0040-4039(97)00846-0 |
| 5. | Aiello, A.; D’Esposito, M.; Fattourusso, E.; Menna, M.; Müller, W. E. G.; Perović-Ottstadt, S.; Schröder, H. C. Bioorg. Med. Chem. 2006, 14, 17–24. doi:10.1016/j.bmc.2005.07.057 |
| 3. | Tsukamoto, S.; Tane, K.; Ohta, T.; Matsunaga, S.; Fusetani, N.; van Soest, R. W. M. J. Nat. Prod. 2001, 64, 1576–1578. doi:10.1021/np010280b |
| 44. | Song, Q.-Y.; Yang, B.-L.; Tian, S.-K. J. Org. Chem. 2007, 72, 5407–5410. doi:10.1021/jo0704558 |
| 2. | Kobayashi, J.; Kanda, F.; Ishibashi, M.; Shigemori, H. J. Org. Chem. 1991, 56, 4574–4576. doi:10.1021/jo00014a052 |
| 6. | Namba, K.; Shinada, T.; Teramoto, T.; Ohfune, Y. J. Am. Chem. Soc. 2000, 122, 10708–10709. doi:10.1021/ja002556s |
| 7. | Wehn, P. M.; Du Bois, J. J. Am. Chem. Soc. 2002, 124, 12950–12951. doi:10.1021/ja028139s |
| 8. | Woo, J. C. S.; MacKay, D. B. Tetrahedron Lett. 2003, 44, 2881–2883. doi:10.1016/S0040-4039(03)00431-3 |
| 9. | Drouin, C.; Woo, J. C. S.; MacKay, D. B.; Lavigne, R. M. A. Tetrahedron Lett. 2004, 45, 7197–7199. doi:10.1016/j.tetlet.2004.08.038 |
| 10. | Kano, T.; Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 2174–2175. doi:10.1021/ja056851u |
| 11. | Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2006, 128, 3928–3930. doi:10.1021/ja060312n |
| 12. | Sibi, M. P.; Stanley, L. M.; Soeta, T. Org. Lett. 2007, 9, 1553–1556. doi:10.1021/ol070364x |
| 13. | Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2007, 9, 1765–1767. doi:10.1021/ol0704789 |
| 14. | Tran, K.; Lombardi, P. J.; Leighton, J. L. Org. Lett. 2008, 10, 3165–3167. doi:10.1021/ol8011869 |
| 15. | Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2010, 12, 2170. doi:10.1021/ol1006339 |
| 16. | Ichikawa, Y.; Okumura, K.; Matsuda, Y.; Hasegawa, T.; Nakamura, M.; Fujimoto, A.; Masuda, T.; Nakano, K.; Kotsuki, H. Org. Biomol. Chem. 2012, 10, 614–622. doi:10.1039/C1OB06559A |
| 17. | Sankar, K.; Rahman, H.; Das, P. P.; Bhimireddy, E.; Sridhar, B.; Mohapatra, D. K. Org. Lett. 2012, 14, 1082–1085. doi:10.1021/ol203466m |
| 18. | Shinada, T.; Oe, K.; Ohfune, Y. Tetrahedron Lett. 2012, 53, 3250–3253. doi:10.1016/j.tetlet.2012.04.042 |
| 19. | Yoshimura, T.; Kinoshita, T.; Yoshioka, H.; Kawabata, T. Org. Lett. 2013, 15, 864–867. doi:10.1021/ol303568f |
| 20. | Nagatomo, M.; Nishiyama, H.; Fujino, H.; Inoue, M. Angew. Chem., Int. Ed. 2015, 127, 1557–1561. doi:10.1002/ange.201410186 |
| 21. | Hashimoto, T.; Maruoka, K. Org. Biomol. Chem. 2008, 6, 829–835. doi:10.1039/B716062C |
| 22. | Ohfune, Y.; Oe, K.; Namba, K.; Shinada, T. Heterocycles 2012, 85, 2617–2649. doi:10.3987/REV-12-746 |
| 38. | Alouane, N.; Boutier, A.; Baron, C.; Vrancken, E.; Mangeney, P. Synthesis 2006, 885–889. doi:10.1055/s-2006-926340 |
| 39. | Broustal, G.; Ariza, X.; Campagne, J.-M.; Garcia, J.; Georges, Y.; Marinetti, A.; Robiette, R. Eur. J. Org. Chem. 2007, 4293–4297. doi:10.1002/ejoc.200700503 |
| 40. | Fraunhoffer, K. J.; White, M. C. J. Am. Chem. Soc. 2007, 129, 7274–7276. doi:10.1021/ja071905g |
| 41. | Amador, M.; Ariza, X.; Garcia, J.; Sevilla, S. Org. Lett. 2002, 4, 4511–4514. doi:10.1021/ol0270428 |
| 42. | Butler, D. C. D.; Inman, G. A.; Alper, H. J. Org. Chem. 2000, 65, 5887–5890. doi:10.1021/jo000608q |
| 43. | Xie, Y.; Yu, K.; Gu, Z. J. Org. Chem. 2014, 79, 1289–1302. doi:10.1021/jo402681z |
| 2. | Kobayashi, J.; Kanda, F.; Ishibashi, M.; Shigemori, H. J. Org. Chem. 1991, 56, 4574–4576. doi:10.1021/jo00014a052 |
| 3. | Tsukamoto, S.; Tane, K.; Ohta, T.; Matsunaga, S.; Fusetani, N.; van Soest, R. W. M. J. Nat. Prod. 2001, 64, 1576–1578. doi:10.1021/np010280b |
| 4. | Jahn, T.; König, G. M.; Wright, A. D.; Wörheide, G.; Reitner, J. Tetrahedron Lett. 1997, 38, 3883–3884. doi:10.1016/S0040-4039(97)00846-0 |
| 5. | Aiello, A.; D’Esposito, M.; Fattourusso, E.; Menna, M.; Müller, W. E. G.; Perović-Ottstadt, S.; Schröder, H. C. Bioorg. Med. Chem. 2006, 14, 17–24. doi:10.1016/j.bmc.2005.07.057 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 2. | Kobayashi, J.; Kanda, F.; Ishibashi, M.; Shigemori, H. J. Org. Chem. 1991, 56, 4574–4576. doi:10.1021/jo00014a052 |
| 29. |
Wang, L.; Menche, D. Angew. Chem. 2012, 124, 9559–9562. doi:10.1002/ange.201203911
Angew. Chem., Int. Ed. 2012, 51, 9425–9427. doi:10.1002/anie.201203911 |
| 32. | Debnar, T.; Dreisigacker, S.; Menche, D. Chem. Commun. 2013, 49, 725–727. doi:10.1039/C2CC37678D |
| 33. | Dieckmann, M.; Menche, D. Org. Lett. 2013, 15, 228–231. doi:10.1021/ol3033303 |
| 34. | Schrempp, M.; Thiede, S.; Herkommer, D.; Gansäuer, A.; Menche, D. Chem. – Eur. J. 2015, 21, 16266–16271. doi:10.1002/chem.201502263 |
| 35. | Thiede, S.; Winterscheid, P. M.; Hartmann, J.; Schnakenburg, G.; Essig, S.; Menche, D. Synthesis 2016, 48, 697–709. doi:10.1055/s-0035-1561278 |
| 36. | Herkommer, D.; Thiede, S.; Wosniok, P. R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. J. Am. Chem. Soc. 2015, 137, 4086–4089. doi:10.1021/jacs.5b01894 |
| 37. | Menche, D.; Arikan, F.; Li, J.; Rudolph, S. Org. Lett. 2007, 9, 267–270. doi:10.1021/ol062715y |
| 6. | Namba, K.; Shinada, T.; Teramoto, T.; Ohfune, Y. J. Am. Chem. Soc. 2000, 122, 10708–10709. doi:10.1021/ja002556s |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 26. | Herkommer, D.; Schmalzbauer, B.; Menche, D. Nat. Prod. Rep. 2014, 31, 456–467. doi:10.1039/C3NP70093C |
| 27. | Tang, B.; Wang, L.; Menche, D. Synlett 2013, 24, 625–629. doi:10.1055/s-0032-1318300 |
| 28. | Wang, L.; Menche, D. J. Org. Chem. 2012, 77, 10811–10823. doi:10.1021/jo302102x |
| 29. |
Wang, L.; Menche, D. Angew. Chem. 2012, 124, 9559–9562. doi:10.1002/ange.201203911
Angew. Chem., Int. Ed. 2012, 51, 9425–9427. doi:10.1002/anie.201203911 |
| 30. |
Wang, L.; Li, P.; Menche, D. Angew. Chem. 2010, 122, 9456–9460. doi:10.1002/ange.201003304
Angew. Chem., Int. Ed. 2010, 49, 9270–9273. doi:10.1002/anie.201003304 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 26. | Herkommer, D.; Schmalzbauer, B.; Menche, D. Nat. Prod. Rep. 2014, 31, 456–467. doi:10.1039/C3NP70093C |
| 27. | Tang, B.; Wang, L.; Menche, D. Synlett 2013, 24, 625–629. doi:10.1055/s-0032-1318300 |
| 28. | Wang, L.; Menche, D. J. Org. Chem. 2012, 77, 10811–10823. doi:10.1021/jo302102x |
| 29. |
Wang, L.; Menche, D. Angew. Chem. 2012, 124, 9559–9562. doi:10.1002/ange.201203911
Angew. Chem., Int. Ed. 2012, 51, 9425–9427. doi:10.1002/anie.201203911 |
| 30. |
Wang, L.; Li, P.; Menche, D. Angew. Chem. 2010, 122, 9456–9460. doi:10.1002/ange.201003304
Angew. Chem., Int. Ed. 2010, 49, 9270–9273. doi:10.1002/anie.201003304 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 6. | Namba, K.; Shinada, T.; Teramoto, T.; Ohfune, Y. J. Am. Chem. Soc. 2000, 122, 10708–10709. doi:10.1021/ja002556s |
| 7. | Wehn, P. M.; Du Bois, J. J. Am. Chem. Soc. 2002, 124, 12950–12951. doi:10.1021/ja028139s |
| 8. | Woo, J. C. S.; MacKay, D. B. Tetrahedron Lett. 2003, 44, 2881–2883. doi:10.1016/S0040-4039(03)00431-3 |
| 9. | Drouin, C.; Woo, J. C. S.; MacKay, D. B.; Lavigne, R. M. A. Tetrahedron Lett. 2004, 45, 7197–7199. doi:10.1016/j.tetlet.2004.08.038 |
| 10. | Kano, T.; Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 2174–2175. doi:10.1021/ja056851u |
| 11. | Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2006, 128, 3928–3930. doi:10.1021/ja060312n |
| 12. | Sibi, M. P.; Stanley, L. M.; Soeta, T. Org. Lett. 2007, 9, 1553–1556. doi:10.1021/ol070364x |
| 13. | Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2007, 9, 1765–1767. doi:10.1021/ol0704789 |
| 14. | Tran, K.; Lombardi, P. J.; Leighton, J. L. Org. Lett. 2008, 10, 3165–3167. doi:10.1021/ol8011869 |
| 15. | Shinada, T.; Ikebe, E.; Oe, K.; Namba, K.; Kawasaki, M.; Ohfune, Y. Org. Lett. 2010, 12, 2170. doi:10.1021/ol1006339 |
| 16. | Ichikawa, Y.; Okumura, K.; Matsuda, Y.; Hasegawa, T.; Nakamura, M.; Fujimoto, A.; Masuda, T.; Nakano, K.; Kotsuki, H. Org. Biomol. Chem. 2012, 10, 614–622. doi:10.1039/C1OB06559A |
| 17. | Sankar, K.; Rahman, H.; Das, P. P.; Bhimireddy, E.; Sridhar, B.; Mohapatra, D. K. Org. Lett. 2012, 14, 1082–1085. doi:10.1021/ol203466m |
| 18. | Shinada, T.; Oe, K.; Ohfune, Y. Tetrahedron Lett. 2012, 53, 3250–3253. doi:10.1016/j.tetlet.2012.04.042 |
| 19. | Yoshimura, T.; Kinoshita, T.; Yoshioka, H.; Kawabata, T. Org. Lett. 2013, 15, 864–867. doi:10.1021/ol303568f |
| 20. | Nagatomo, M.; Nishiyama, H.; Fujino, H.; Inoue, M. Angew. Chem., Int. Ed. 2015, 127, 1557–1561. doi:10.1002/ange.201410186 |
| 21. | Hashimoto, T.; Maruoka, K. Org. Biomol. Chem. 2008, 6, 829–835. doi:10.1039/B716062C |
| 22. | Ohfune, Y.; Oe, K.; Namba, K.; Shinada, T. Heterocycles 2012, 85, 2617–2649. doi:10.3987/REV-12-746 |
| 54. | Goulaouic-Dubois, C.; Guggisberg, A.; Hesse, M. Tetrahedron 1995, 51, 12573–12582. doi:10.1016/0040-4020(95)00811-L |
| 55. | Knowles, H. S.; Parsons, A. F.; Pettifer, R. M.; Rickling, S. Tetrahedron 2000, 56, 979–988. doi:10.1016/S0040-4020(00)00025-9 |
| 23. | Urban, S.; de Almeida Leone, P.; Carroll, A. R.; Fechner, G. A.; Smith, J.; Hooper, J. N. A.; Quinn, R. J. J. Org. Chem. 1999, 64, 731–735. doi:10.1021/jo981034g |
| 24. | Kobayashi, J.; Inaba, K.; Tsuda, M. Tetrahedron 1997, 53, 16679–16682. doi:10.1016/S0040-4020(97)10097-7 |
| 25. | Kato, T.; Shizuri, Y.; Izumida, H.; Yokoyama, A.; Endo, M. Tetrahedron Lett. 1995, 36, 2133–2136. doi:10.1016/0040-4039(95)00194-H |
| 6. | Namba, K.; Shinada, T.; Teramoto, T.; Ohfune, Y. J. Am. Chem. Soc. 2000, 122, 10708–10709. doi:10.1021/ja002556s |
| 56. | Pappo, R.; Allen, D. S., Jr.; Lemieux, R. U.; Johnson, W. S. J. Org. Chem. 1956, 21, 478–479. doi:10.1021/jo01110a606 |
| 57. | Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett. 2004, 6, 3217–3219. doi:10.1021/ol0400342 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 39. | Broustal, G.; Ariza, X.; Campagne, J.-M.; Garcia, J.; Georges, Y.; Marinetti, A.; Robiette, R. Eur. J. Org. Chem. 2007, 4293–4297. doi:10.1002/ejoc.200700503 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
| 50. | Herrington, P. M.; Owen, C. E.; Gage, J. R. Org. Process Res. Dev. 2001, 5, 80–83. doi:10.1021/op0002949 |
| 51. | Formentín, P.; Gimeno, N.; Steinke, J. H. G.; Vilar, R. J. Org. Chem. 2005, 70, 8235–8238. doi:10.1021/jo051120y |
| 49. | Li, H.; Hong, J.-H. Bull. Korean Chem. Soc. 2008, 29, 847–850. doi:10.5012/bkcs.2008.29.4.847 |
| 46. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 47. | Tang, T. P.; Volkman, S. K.; Ellman, J. A. J. Org. Chem. 2001, 66, 8772–8778. doi:10.1021/jo0156868 |
| 48. | Yendapally, R.; Lee, R. E. Bioorg. Med. Chem. Lett. 2008, 18, 1607–1611. doi:10.1016/j.bmcl.2008.01.065 |
| 46. | Cogan, D. A.; Liu, G.; Ellman, J. Tetrahedron 1999, 55, 8883–8904. doi:10.1016/S0040-4020(99)00451-2 |
| 47. | Tang, T. P.; Volkman, S. K.; Ellman, J. A. J. Org. Chem. 2001, 66, 8772–8778. doi:10.1021/jo0156868 |
| 48. | Yendapally, R.; Lee, R. E. Bioorg. Med. Chem. Lett. 2008, 18, 1607–1611. doi:10.1016/j.bmcl.2008.01.065 |
| 39. | Broustal, G.; Ariza, X.; Campagne, J.-M.; Garcia, J.; Georges, Y.; Marinetti, A.; Robiette, R. Eur. J. Org. Chem. 2007, 4293–4297. doi:10.1002/ejoc.200700503 |
| 31. | Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494–4497. doi:10.1021/ol101755m |
© 2016 Bretzke et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)