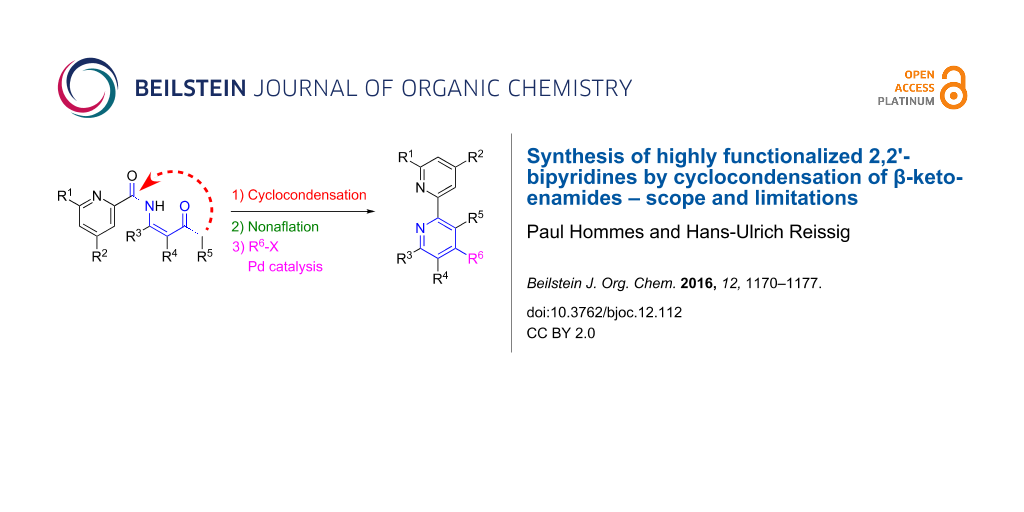
Abstract
The scope of a flexible route to unsymmetrically functionalized bipyridines is described. Starting from 1,3-diketones 1a–e, the corresponding β-ketoenamines 2a–e were converted into different β-ketoenamides 3a–g by N-acylation with 2-pyridinecarboxylic acid derivatives. These β-ketoenamides were treated with a mixture of TMSOTf and Hünig’s base to promote the cyclocondensation to 4-hydroxypyridine derivatives. Their immediate O-nonaflation employing nonafluorobutanesulfonyl fluoride provided the expected 4-nonafloxy-substituted bipyridine derivatives 5a–g in moderate to good overall yields. The bipyridyl nonaflates are excellent precursors for palladium-catalyzed reactions as demonstrated by representative Suzuki and Sonogashira couplings. Thus, a library of specifically substituted bipyridine derivatives was generated, showing the versatility of the simple 1,3-diketone-based approach to this important class of ligands.

Graphical Abstract
Introduction
In 2009 we reported on a new pyridine synthesis starting from symmetrically substituted 1,3-diketones 1a and 1b, respectively, that were converted into the corresponding β-ketoenamines and subsequently by N-acylation into β-ketoenamides such as 3a or 3b [1]. Their cyclocondensation followed by O-alkylation or O-nonaflation led to functionalized pyridine derivatives. Scheme 1 illustrates this sequence with the synthesis of two 2,2´-bipyridine derivatives 4a or 5b that were obtained by employing picolinic acid chloride for the N-acylations followed by O-methylation with methyl iodide or by O-nonaflation using nonafluorobutanesulfonyl fluoride (NfF) as sulfonylating reagent. In subsequent publications we could demonstrate that this method can be used to synthesize a variety of functionalized pyridine derivatives, 2,2´-bipyridines or terpyridines [2-4]. An alternative entry to the crucial β-ketoenamides via a Blaise reaction was also developed [5], however, only a limited number of examples for the preparation of bipyridine derivatives have been reported so far. Hence we describe here additional experiments leading to this important class of compounds, in order to define scope and limitations of this reaction sequence. For this purpose we prepared other β-ketoenamides, studied their cyclocondensation reactions and used the present functionalities for subsequent reactions, in particular palladium-catalyzed coupling reactions.
![[1860-5397-12-112-i1]](/bjoc/content/inline/1860-5397-12-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of highly functionalized 2,2'-bipyridines 4a and 5b from symmetrical 1,3-diketones 1a and 1b; TMSOTf = trimethylsilyl trifluoromethanesulfonate; DIPEA = N,N-diisopropylethylamine; NfF = nonafluorobutanesulfonyl fluoride.
Scheme 1: Synthesis of highly functionalized 2,2'-bipyridines 4a and 5b from symmetrical 1,3-diketones 1a and ...
2,2´-Bipyridines are a very important class of ligands employed in catalysis, supramolecular chemistry or materials science and hence a large number of specifically substituted derivatives is known (for reviews and selected recent syntheses or applications see [6-20]). Due to the flexible and modular nature of our approach to this compound class the design of new derivatives is possible thus supplementing the existing collection of available 2,2´-bipyridines.
Results and Discussion
First we prepared the corresponding β-ketoenamines 2 required for the N-acylation step (Scheme 2). The known compounds 2a and 2b are available in excellent yields from the 1,3-diketones 1a and 1b employing aqueous ammonia in the presence of silica gel [21]. Compound 2c was prepared from 1c in 87% yield with ammonium formate [22,23]. Interestingly, treatment of 1c with aqueous ammonia/silica gel failed to give 2c, deacetylation to 1-phenylpropan-2-one (76% yield) was observed instead. For the amination of benzyl-substituted diketone 1d both of the above utilized methods failed to provide the desired compound 2d. While treatment with ammonium formate exclusively leads to deacetylation of the starting material to 4-phenylbutan-2-one, the reaction with aqueous ammonia/silica gel provided a mixture of the desired compound 2d and the deacetylation product. In both methods slightly acidic reagents are employed, in contrast strictly basic conditions provided by the reaction of 1d with a 7 M solution of ammonia in methanol allowed preparing the desired β-ketoenamine 2d in a reasonable yield. The reaction of compound 1e with NH3/SiO2 also mainly induced decomposition by deacetylation but the desired β-ketoenamine 2e could be prepared in 70% yield using ammonium acetate in toluene [24]. The reaction of 1e with ammonium formate merely provided 2e in 42% yield.
![[1860-5397-12-112-i2]](/bjoc/content/inline/1860-5397-12-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of β-ketoenamines 2c–e and of β-ketoenamides 3c–h.
Scheme 2: Synthesis of β-ketoenamines 2c–e and of β-ketoenamides 3c–h.
The corresponding β-ketoenamides containing pyridine moieties at the acylated nitrogen were prepared by standard methods with the respective acid chlorides that furnished compounds 3c and 3d in good yields. For the synthesis of compound 3e it was advantageous to employ the benzotriazole derivative 2-PyCOBt [25,26]. During an attempt to prepare β-ketoenamide 3a by in situ generation of picolinic acid chloride with thionyl chloride under heating and by amide formation of the resulting product with 2a we isolated not only the expected compound 3a but as major component the meta-chlorinated compound 3f. This subsequent chlorination of picolinic acid chloride is known from the literature to proceed under enforced conditions [27,28]. On the other hand, a selective synthesis of 3a is possible in 73% yield by coupling of β-enaminoketone 2a with picolinic acid under assistance of BOP as reagent (see Supporting Information File 1). The in situ generation of the acid chloride with thionyl chloride was possible without the undesired chlorination with the more electron-deficient 6-methoxycarbonyl-substituted 2-pyridine carboxylic acid delivering with 2a the desired β-ketoenamide 3g with excellent efficacy.
We also examined an alternative approach to β-ketoenamides by the addition of carboxylic acid amides to alkynones. This approach can potentially lead to products that are formally derived from unsymmetrically substituted 1,3-dicarbonyl compounds. The best result was obtained with picolinic acid amide and but-3-yn-2-one (as an equivalent of 3-oxobutanal) using methanol as solvent in a sealed tube [29]. Disappointingly, the maximum yield for compound 3h was only 22%. Many attempts to improve this result – including gold catalysis [30] – either led to lower yields or no conversion at all [31]. We have to conclude that this approach to β-ketoenamides is not efficient.
With N-pyridyl-substituted β-ketoenamides 3a–h in hand, we investigated their cyclocondensations to the corresponding 2,2´-bipyridine derivatives 5a–h (Table 1). As in our preliminary experiments the use of trimethylsilyl trifluoromethanesulfonate and Hünig’s base (DIPEA) gave the best results if the mixture was heated for 3 d in 1,2-dichloroethane at 90 °C under fairly high dilution (ca. 0.04 M). Higher concentrations as reported earlier [1] gave inferior results, probably due to increased amounts of intermolecular reactions leading to oligomeric side products. The resulting primary condensation products contain a 4-hydroxy group in the newly formed pyridine ring that also exists as 4-pyridone tautomer making the identification and purification at this stage inconvenient. We therefore removed all volatile components from the crude products of the cyclocondensation step and subjected the unpurified compounds to the O-nonaflation procedure. Treatment with an excess of sodium hydride in THF followed by reaction with nonafluorobutanesulfonyl fluoride (NfF) afforded the desired 2,2´-bipyridine derivatives 5a–g containing the 4-nonafloxy substituent in moderate to good overall yields. This functional group fixes the tautomeric structure and makes the products less polar which allowed smooth isolation, purification and identification. An additional advantage of this substituent is its ability to allow transition-metal-catalyzed coupling reactions [32]. Entries 1–7 of Table 1 demonstrate that quite differently substituted 2,2´-bipyridine derivatives are accessible by this method. Only precursor 3h that should lead to compound 5h with all substituents R1–R5 being hydrogen could not be isolated. We assume that the missing bulk of substituents led to undesired side reactions, e.g., deacylation or oligomerization processes.
Table 1: Attempted TMSOTf/DIPEA-promoted cyclocondensations of β-ketoenamides 3a–h and subsequent O-nonaflation leading to 2,2'-bipyridines 5a–h.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-112-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry |
β-Keto-
enamide |
R1 | R2 | R3 | R4 | R5 |
2,2'-Bipyrid-4-yl
nonaflate (yield)a |
|---|---|---|---|---|---|---|---|
| 1 | 3a | H | H | Me | H | H | 5a (75%) |
| 2 | 3b | H | H | Et | H | Me | 5b (51%)b |
| 3 | 3c | CO2Me | H | Me | Ph | H | 5c (86%) |
| 4 | 3d | H | H | Me | Bn | H | 5d (54%) |
| 5c | 3e | H | H | CO2Et | H | H | 5e (52%) |
| 6 | 3f | H | Cl | Me | H | H | 5f (66%) |
| 7 | 3g | CO2Me | H | Me | H | H | 5g (51%) |
| 8 | 3h | H | H | H | H | H | 5h –d |
aYield over two steps. bExample taken from ref. [1]. cNf2O was used instead of NfF. dNo defined products isolated.
The development of the discussed pyridine syntheses by cyclocondensation reactions of β-ketoenamides was inspired by a serendipitously discovered three-component reaction of lithiated alkoxyallenes, nitriles and carboxylic acids [33-40]. These components form α-alkoxy-substituted β-ketoenamides as precursors of highly substituted pyridine derivatives. We therefore tried to independently prepare β-ketoenamides with this substitution pattern starting from simple 1,3-diketones such as 1a. An oxidative coupling of acetylacetone 1a employing iodosylbenzene in the presence of boron trifluoride in methanol [41] followed by the standard amination procedure indeed furnished β-enaminoketone 2i in moderate yield (Scheme 3). This intermediate could be N-acylated via its potassium salt with activated picolinic acid to furnish the desired α-methoxy-β-ketoenamide 3i in 50% yield. This sequence demonstrates that this type of β-ketoenamide precursor is also available by this oxidative method; in this case the direct preparation of 3i by the multicomponent route employing methoxyallene was similarly efficient (22% yield, [39]). It has been earlier demonstrated that 3i can be cyclized and O-methylated to the corresponding bipyridine derivative 4i in 53% yield (see Scheme 4) [39].
![[1860-5397-12-112-i3]](/bjoc/content/inline/1860-5397-12-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of α-methoxy-β-ketoenamine 2i, its N-acylation to 3i and the reaction of β-ketoenamide 3a with iodosylbenzene to 5-acetyloxazole 6.
Scheme 3: Synthesis of α-methoxy-β-ketoenamine 2i, its N-acylation to 3i and the reaction of β-ketoenamide 3a...
When we tried to employ the oxidative methoxylation to β-ketoenamide 3a the expected methoxy-substituted product 3i was not formed. Instead, we isolated the trisubstituted oxazole derivative 6 in 55% yield. The scope of this reaction has not been studied by us, however it is known in the literature that enamides can undergo related oxidative cyclizations to oxazoles in the presence of the reagent cocktail employed or under related reaction conditions [41-45]. In this context we want to notice that our approach to alkoxy-substituted β-ketoenamides via alkoxyallenes also allowed the synthesis of 5-acetyloxazoles with high flexibility, if the acid-labile trimethylsilylethyl group was introduced via the alkoxyallene [46,47].
As mentioned above β-ketoenamide 3i is a good precursor for 2,2´-bipyridine derivative 4i, but Scheme 4 also reveals that this intermediate could be converted into the expected pyrimidine derivative 7 by treatment with ammonium acetate [48-50] or into pyrimidine N-oxide 8 by employing hydroxylamine hydrochloride [51,52]. These products of simple condensation reactions may be regarded as azabipyridine derivatives and their synthesis demonstrates the versatility of β-ketoenamide such as 3i for the synthesis of highly functionalized heterocycles.
![[1860-5397-12-112-i4]](/bjoc/content/inline/1860-5397-12-112-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclizations of β-ketoenamide 3i leading to 2,2´-bipyridine derivative 4i or to the related 2-(2-pyridyl)pyrimidine 7 or the corresponding N-oxide 8.
Scheme 4: Cyclizations of β-ketoenamide 3i leading to 2,2´-bipyridine derivative 4i or to the related 2-(2-py...
The 2,2´-bipyridyl nonaflates prepared by the methods described above are excellent and versatile precursors for transition-metal-catalyzed coupling reactions [32,53-57]. Standard conditions for a Suzuki coupling smoothly transform compound 5a and a boronic acid derivative into the expected 4-tolyl-substituted derivative 9 and the higher substituted precursor 5b was converted into compound 10 by employing an acetyl-substituted boronic acid derivate in very good yield (Scheme 5). Compound 10 was prepared in order to investigate its capability to form a C3-symmetric star-shaped product by cyclocondensation of the acetyl group. This type of aldol condensation that generates a new benzene ring from three acetyl groups was successfully applied in our group in other cases delivering compounds whose 2D self-assembly at highly ordered pyrolytic graphite was studied by STM methods [58,59]. However, in the case of compound 10 no full conversion to the desired star-shaped compound was observed under the applied reaction conditions (SiCl4, EtOH) [60,61], very likely due to the insolubility of the intermediates involved in this multistep process.
![[1860-5397-12-112-i5]](/bjoc/content/inline/1860-5397-12-112-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Suzuki-couplings of 2,2'-bipyrid-4-yl nonaflates 5a and 5b to compounds 9 and 10.
Scheme 5: Suzuki-couplings of 2,2'-bipyrid-4-yl nonaflates 5a and 5b to compounds 9 and 10.
The chloro-substituted 2,2´-bipyridine derivative 5g – available due to the accidental chlorination of the picolinic acid precursor (Scheme 2) – offered the option to perform two subsequent coupling reactions in a controlled fashion. In the first step, the more reactive nonaflate moiety of 5g was efficiently substituted by a phenyl group under standard Suzuki reaction conditions (Scheme 6). The untouched chloro substituent in the second pyridine ring was then replaced by another aryl substituent leading to product 12 in 54% yield. Alternatively, a Sonogashira reaction with (cyclopropyl)ethyne transformed intermediate 11 into the alkynyl-substituted 2,2´-bipyridine derivative 13. Again the yield of this second coupling step is only moderate since the chloro compound 11 is not very reactive; in both cases even after two days reaction time ca. 50% of the precursor was re-isolated. Nevertheless, the examples in Scheme 5 and Scheme 6 clearly demonstrate that by suitable subsequent reactions our library of unsymmetrically substituted 2,2´-bipyridine derivatives 5 can easily be enlarged in a flexible manner.
![[1860-5397-12-112-i6]](/bjoc/content/inline/1860-5397-12-112-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Palladium-catalyzed couplings of chloro-substituted 2,2'-bipyrid-4-yl nonaflate 5g leading to compounds 11, 12 and 13.
Scheme 6: Palladium-catalyzed couplings of chloro-substituted 2,2'-bipyrid-4-yl nonaflate 5g leading to compo...
Conclusion
In this report we could show that the scope of the cyclocondensation reaction of β-ketoenamides 3 leading to unsymmetrically substituted bipyridine derivatives 5 is fairly broad and compatible with various functional groups. Only substrates such as 3h having no “superfluous” substituents did not provide the expected bipyridine skeleton. Substrates 3 with higher substitution degree provided the expected bipyridine derivatives that were directly converted into the corresponding nonafloxy-substituted compounds 5 in good overall yield. These are excellent precursors for palladium-catalyzed coupling reactions as exemplified by the synthesis of compounds such as 9–13. By an attempt to prepare the methoxy-substituted β-ketoenamide 3i by oxidative methoxylation of compound 3a we unexpectedly observed the formation of the 5-acetyl-substituted oxazole derivative 6. However, precursor 3i was smoothly available from acetylacetone 1a. It could smoothly be transformed into the desired bipyridine derivative 4i, but also into the related pyrimidine and pyrimidine N-oxide derivatives 7 and 8. Altogether, we could confirm that our approach to functionalized pyridine derivatives by cyclocondensation of β-ketoenamides is also a very versatile and flexible method for the synthesis of unsymmetrically substituted bipyridine derivatives or related heterocyclic compounds that are not accessible by alternative methods.
Supporting Information
| Supporting Information File 1: General information, all experimental procedures and analytical data as well as copies of NMR spectra of all compounds. | ||
| Format: PDF | Size: 4.0 MB | Download |
References
-
Dash, J.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939
Return to citation in text: [1] [2] [3] -
Eidamshaus, C.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 6056–6069. doi:10.1002/ejoc.201100681
Return to citation in text: [1] -
Hommes, P.; Berlin, S.; Reissig, H.-U. Synthesis 2013, 45, 3288–3294. doi:10.1055/s-0033-1338548
Return to citation in text: [1] -
Hommes, P.; Fischer, C.; Lindner, C.; Zipse, H.; Reissig, H.-U. Angew. Chem., Int. Ed. 2014, 53, 7647–7651. doi:10.1002/anie.201403403
Angew. Chem. 2014, 126, 7778–7782. doi:10.1002/ange.201403403
Return to citation in text: [1] -
Hommes, P.; Jungk, P.; Reissig, H.-U. Synlett 2011, 2311–2314. doi:10.1055/s-0030-1260304
Return to citation in text: [1] -
Chelucci, G.; Thummel, R. P. Chem. Rev. 2002, 102, 3129–3170. doi:10.1021/cr0101914
Return to citation in text: [1] -
Kaes, C.; Katz, A.; Hosseini, M. W. Chem. Rev. 2000, 100, 3553–3590. doi:10.1021/cr990376z
Return to citation in text: [1] -
Schubert, U. S.; Eschbaumer, C. Angew. Chem., Int. Ed. 2002, 41, 2892–2926. doi:10.1002/1521-3773(20020816)41:16<2892::AID-ANIE2892>3.0.CO;2-6
Angew. Chem. 2002, 114, 3016–3050. doi:10.1002/1521-3757(20020816)114:16<3016::AID-ANGE3016>3.0.CO;2-5
Return to citation in text: [1] -
Fletcher, N. C. J. Chem. Soc., Perkin Trans. 1 2002, 1831–1842. doi:10.1039/b204272j
Return to citation in text: [1] -
Newkome, G. R.; Patri, A. K.; Holder, E.; Schubert, U. S. Eur. J. Org. Chem. 2004, 235–254. doi:10.1002/ejoc.200300399
Return to citation in text: [1] -
Ye, B.-H.; Tong, M.-L.; Chen, X.-M. Coord. Chem. Rev. 2005, 249, 545–565. doi:10.1016/j.ccr.2004.07.006
Return to citation in text: [1] -
Hapke, M.; Brandt, L.; Lützen, A. Chem. Soc. Rev. 2008, 37, 2782–2797. doi:10.1039/b810973g
Return to citation in text: [1] -
Gütz, C.; Lützen, A. Synthesis 2010, 85–90. doi:10.1055/s-0029-1217082
Return to citation in text: [1] -
Bozic-Weber, B.; Constable, E. C.; Housecroft, C. E.; Kopecky, P.; Neuburger, M.; Zampese, J. A. Dalton Trans. 2011, 40, 12584–12594. doi:10.1039/c1dt11052g
Return to citation in text: [1] -
Duric, S.; Tzschucke, C. C. Org. Lett. 2011, 13, 2310–2313. doi:10.1021/ol200565u
Return to citation in text: [1] -
Okamoto, S. Heterocycles 2012, 85, 1579–1602. doi:10.3987/REV-12-737
Return to citation in text: [1] -
Duric, S.; Sypaseuth, F. D.; Hoof, S.; Svensson, E.; Tzschucke, C. C. Chem. – Eur. J. 2013, 19, 17456–17463. doi:10.1002/chem.201302118
Return to citation in text: [1] -
Kremer, C.; Schnakenburg, G.; Lützen, A. Beilstein J. Org. Chem. 2014, 10, 814–824. doi:10.3762/bjoc.10.77
Return to citation in text: [1] -
Guo, Z.; Tong, W.-L.; Chan, M. C. W. Chem. Commun. 2014, 50, 1711–1714. doi:10.1039/c3cc47150k
Return to citation in text: [1] -
Renfrew, A. K.; Bryce, N. S.; Hambley, T. Chem. – Eur. J. 2015, 21, 15224–15234. doi:10.1002/chem.201502702
Return to citation in text: [1] -
Gao, Y.; Zhang, Q.; Xu, J. Synth. Commun. 2004, 34, 909–916. doi:10.1081/scc-120028364
Return to citation in text: [1] -
Ji, Y.; Trenkle, W. C.; Vowles, J. V. Org. Lett. 2006, 8, 1161–1163. doi:10.1021/ol053164z
Return to citation in text: [1] -
Li, X.; Du, Y.; Liang, Z.; Li, X.; Pan, Y.; Zhao, K. Org. Lett. 2009, 11, 2643–2646. doi:10.1021/ol9006663
Return to citation in text: [1] -
Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron 1987, 43, 235–242. doi:10.1016/S0040-4020(01)89949-X
Return to citation in text: [1] -
Katritzky, A. R.; He, H.-Y.; Suzuki, K. J. Org. Chem. 2000, 65, 8210–8213. doi:10.1021/jo000792f
Return to citation in text: [1] -
Katritzky, A. R.; Suzuki, K.; Wang, Z. Synlett 2005, 1656–1665. doi:10.1055/s-2005-871551
Return to citation in text: [1] -
Mosher, H. S.; Look, M. J. Org. Chem. 1955, 20, 283–286. doi:10.1021/jo01121a002
Return to citation in text: [1] -
Bankston, D.; Dumas, J.; Natero, R.; Riedl, B.; Monahan, M.-K.; Sibley, R. Org. Process Res. Dev. 2002, 6, 777–781. doi:10.1021/op020205n
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G.; Filisti, E. Org. Lett. 2008, 10, 2629–2632. doi:10.1021/ol800518j
Return to citation in text: [1] -
Kobayashi, S.; Kakumoto, K.; Sugiura, M. Org. Lett. 2002, 4, 1319–1322. doi:10.1021/ol0256163
Return to citation in text: [1] -
Hommes, P. Ph.D. Thesis, Freie Universität Berlin, 2013.
Return to citation in text: [1] -
Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566
Return to citation in text: [1] [2] -
Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1844. doi:10.1351/pac-con-09-09-06
Return to citation in text: [1] -
Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem. – Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322
Return to citation in text: [1] -
Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398
Return to citation in text: [1] -
Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/b925468d
Return to citation in text: [1] [2] [3] -
Eidamshaus, C.; Kumar, R.; Bera, M. K.; Reissig, H.-U. Beilstein J. Org. Chem. 2011, 7, 962–975. doi:10.3762/bjoc.7.108
Return to citation in text: [1] -
Moriarty, R. M.; Vaid, R. K.; Ravikumar, V. T.; Vaid, B. K.; Hopkins, T. E. Tetrahedron 1988, 44, 1603–1607. doi:10.1016/S0040-4020(01)86720-X
Return to citation in text: [1] [2] -
Zheng, Y.; Li, X.; Ren, C.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. J. Org. Chem. 2012, 77, 10353–10361. doi:10.1021/jo302073e
Return to citation in text: [1] -
Hempel, C.; Nachtsheim, B. J. Synthesis 2013, 24, 2119–2123. doi:10.1055/s-0033-1339491
Return to citation in text: [1] -
Zheng, M.; Huang, L.; Huang, H.; Li, X.; Wu, W.; Jiang, H. Org. Lett. 2014, 16, 5906–5909. doi:10.1021/ol502916a
Return to citation in text: [1] -
Guo, W.-C.; Hu, F.; Huo, Y.-M.; Chang, H.-H.; Li, X.; Wie, W.-L. Org. Lett. 2015, 17, 3914–3917. doi:10.1021/acs.orglett.5b01933
Return to citation in text: [1] -
Lechel, T.; Lentz, D.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386
Return to citation in text: [1] -
Lechel, T.; Gerhard, M.; Trawny, D.; Brusilowskij, B.; Schefzig, L.; Zimmer, R.; Rabe, J. P.; Lentz, D.; Schalley, C. A.; Reissig, H.-U. Chem. – Eur. J. 2011, 17, 7480–7491. doi:10.1002/chem.201100382
Return to citation in text: [1] -
Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220
Return to citation in text: [1] -
Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056
Return to citation in text: [1] -
Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787
Return to citation in text: [1] -
Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088
Return to citation in text: [1] -
Bera, M. K.; Domínguez, M.; Hommes, P.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 394–404. doi:10.3762/bjoc.10.37
Return to citation in text: [1] -
Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem. – Eur. J. 2011, 17, 11838–11843. doi:10.1002/chem.201101739
Return to citation in text: [1] -
Eidamshaus, C.; Hommes, P.; Reissig, H.-U. Synlett 2012, 23, 1670–1674. doi:10.1055/s-0031-1290398
Return to citation in text: [1] -
Gholap, S. L.; Hommes, P.; Neuthe, K.; Reissig, H.-U. Org. Lett. 2013, 15, 318–321. doi:10.1021/ol303231c
Return to citation in text: [1] -
Bera, M. K.; Gholap, S. L.; Hommes, P.; Neuthe, K.; Trawny, D.; Rabe, J. P.; Lentz, D.; Zimmer, R.; Reissig, H.-U. Adv. Synth. Catal. 2013, 355, 3463–3474. doi:10.1002/adsc.201300613
Return to citation in text: [1] -
Podolan, G.; Hettmanczyk, L.; Hommes, P.; Sarkar, B.; Reissig, H.-U. Eur. J. Org. Chem. 2015, 7317–7323. doi:10.1002/ejoc.201501163
Return to citation in text: [1] -
Trawny, D.; Vandromme, L.; Rabe, J. P.; Reissig, H. U. Eur. J. Org. Chem. 2014, 4985–4992. doi:10.1002/ejoc.201402178
Return to citation in text: [1] -
Dash, J.; Trawny, D.; Rabe, J. P.; Reissig, H. U. Synlett 2015, 26, 1486–1489. doi:10.1055/s-0034-1380716
Return to citation in text: [1] -
Kotha, S.; Kashinath, D.; Lahiri, K.; Sunoj, R. B. Eur. J. Org. Chem. 2004, 4003–4013. doi:10.1002/ejoc.200400257
Return to citation in text: [1] -
Cao, X.-Y.; Liu, X.-H.; Zhou, X.-H.; Zhang, Y.; Jiang, Y.; Cao, Y.; Cui, Y.-X.; Pei, J. J. Org. Chem. 2004, 69, 6050–6058. doi:10.1021/jo049268p
Return to citation in text: [1]
| 1. | Dash, J.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939 |
| 21. | Gao, Y.; Zhang, Q.; Xu, J. Synth. Commun. 2004, 34, 909–916. doi:10.1081/scc-120028364 |
| 1. | Dash, J.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939 |
| 6. | Chelucci, G.; Thummel, R. P. Chem. Rev. 2002, 102, 3129–3170. doi:10.1021/cr0101914 |
| 7. | Kaes, C.; Katz, A.; Hosseini, M. W. Chem. Rev. 2000, 100, 3553–3590. doi:10.1021/cr990376z |
| 8. |
Schubert, U. S.; Eschbaumer, C. Angew. Chem., Int. Ed. 2002, 41, 2892–2926. doi:10.1002/1521-3773(20020816)41:16<2892::AID-ANIE2892>3.0.CO;2-6
Angew. Chem. 2002, 114, 3016–3050. doi:10.1002/1521-3757(20020816)114:16<3016::AID-ANGE3016>3.0.CO;2-5 |
| 9. | Fletcher, N. C. J. Chem. Soc., Perkin Trans. 1 2002, 1831–1842. doi:10.1039/b204272j |
| 10. | Newkome, G. R.; Patri, A. K.; Holder, E.; Schubert, U. S. Eur. J. Org. Chem. 2004, 235–254. doi:10.1002/ejoc.200300399 |
| 11. | Ye, B.-H.; Tong, M.-L.; Chen, X.-M. Coord. Chem. Rev. 2005, 249, 545–565. doi:10.1016/j.ccr.2004.07.006 |
| 12. | Hapke, M.; Brandt, L.; Lützen, A. Chem. Soc. Rev. 2008, 37, 2782–2797. doi:10.1039/b810973g |
| 13. | Gütz, C.; Lützen, A. Synthesis 2010, 85–90. doi:10.1055/s-0029-1217082 |
| 14. | Bozic-Weber, B.; Constable, E. C.; Housecroft, C. E.; Kopecky, P.; Neuburger, M.; Zampese, J. A. Dalton Trans. 2011, 40, 12584–12594. doi:10.1039/c1dt11052g |
| 15. | Duric, S.; Tzschucke, C. C. Org. Lett. 2011, 13, 2310–2313. doi:10.1021/ol200565u |
| 16. | Okamoto, S. Heterocycles 2012, 85, 1579–1602. doi:10.3987/REV-12-737 |
| 17. | Duric, S.; Sypaseuth, F. D.; Hoof, S.; Svensson, E.; Tzschucke, C. C. Chem. – Eur. J. 2013, 19, 17456–17463. doi:10.1002/chem.201302118 |
| 18. | Kremer, C.; Schnakenburg, G.; Lützen, A. Beilstein J. Org. Chem. 2014, 10, 814–824. doi:10.3762/bjoc.10.77 |
| 19. | Guo, Z.; Tong, W.-L.; Chan, M. C. W. Chem. Commun. 2014, 50, 1711–1714. doi:10.1039/c3cc47150k |
| 20. | Renfrew, A. K.; Bryce, N. S.; Hambley, T. Chem. – Eur. J. 2015, 21, 15224–15234. doi:10.1002/chem.201502702 |
| 33. | Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1844. doi:10.1351/pac-con-09-09-06 |
| 34. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem. – Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 35. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 36. | Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398 |
| 37. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 38. | Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183 |
| 39. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/b925468d |
| 40. | Eidamshaus, C.; Kumar, R.; Bera, M. K.; Reissig, H.-U. Beilstein J. Org. Chem. 2011, 7, 962–975. doi:10.3762/bjoc.7.108 |
| 5. | Hommes, P.; Jungk, P.; Reissig, H.-U. Synlett 2011, 2311–2314. doi:10.1055/s-0030-1260304 |
| 1. | Dash, J.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939 |
| 2. | Eidamshaus, C.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 6056–6069. doi:10.1002/ejoc.201100681 |
| 3. | Hommes, P.; Berlin, S.; Reissig, H.-U. Synthesis 2013, 45, 3288–3294. doi:10.1055/s-0033-1338548 |
| 4. |
Hommes, P.; Fischer, C.; Lindner, C.; Zipse, H.; Reissig, H.-U. Angew. Chem., Int. Ed. 2014, 53, 7647–7651. doi:10.1002/anie.201403403
Angew. Chem. 2014, 126, 7778–7782. doi:10.1002/ange.201403403 |
| 32. | Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566 |
| 27. | Mosher, H. S.; Look, M. J. Org. Chem. 1955, 20, 283–286. doi:10.1021/jo01121a002 |
| 28. | Bankston, D.; Dumas, J.; Natero, R.; Riedl, B.; Monahan, M.-K.; Sibley, R. Org. Process Res. Dev. 2002, 6, 777–781. doi:10.1021/op020205n |
| 30. | Kobayashi, S.; Kakumoto, K.; Sugiura, M. Org. Lett. 2002, 4, 1319–1322. doi:10.1021/ol0256163 |
| 25. | Katritzky, A. R.; He, H.-Y.; Suzuki, K. J. Org. Chem. 2000, 65, 8210–8213. doi:10.1021/jo000792f |
| 26. | Katritzky, A. R.; Suzuki, K.; Wang, Z. Synlett 2005, 1656–1665. doi:10.1055/s-2005-871551 |
| 24. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron 1987, 43, 235–242. doi:10.1016/S0040-4020(01)89949-X |
| 22. | Ji, Y.; Trenkle, W. C.; Vowles, J. V. Org. Lett. 2006, 8, 1161–1163. doi:10.1021/ol053164z |
| 23. | Li, X.; Du, Y.; Liang, Z.; Li, X.; Pan, Y.; Zhao, K. Org. Lett. 2009, 11, 2643–2646. doi:10.1021/ol9006663 |
| 29. | Cacchi, S.; Fabrizi, G.; Filisti, E. Org. Lett. 2008, 10, 2629–2632. doi:10.1021/ol800518j |
| 39. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/b925468d |
| 41. | Moriarty, R. M.; Vaid, R. K.; Ravikumar, V. T.; Vaid, B. K.; Hopkins, T. E. Tetrahedron 1988, 44, 1603–1607. doi:10.1016/S0040-4020(01)86720-X |
| 39. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/b925468d |
| 60. | Kotha, S.; Kashinath, D.; Lahiri, K.; Sunoj, R. B. Eur. J. Org. Chem. 2004, 4003–4013. doi:10.1002/ejoc.200400257 |
| 61. | Cao, X.-Y.; Liu, X.-H.; Zhou, X.-H.; Zhang, Y.; Jiang, Y.; Cao, Y.; Cui, Y.-X.; Pei, J. J. Org. Chem. 2004, 69, 6050–6058. doi:10.1021/jo049268p |
| 32. | Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566 |
| 53. | Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem. – Eur. J. 2011, 17, 11838–11843. doi:10.1002/chem.201101739 |
| 54. | Eidamshaus, C.; Hommes, P.; Reissig, H.-U. Synlett 2012, 23, 1670–1674. doi:10.1055/s-0031-1290398 |
| 55. | Gholap, S. L.; Hommes, P.; Neuthe, K.; Reissig, H.-U. Org. Lett. 2013, 15, 318–321. doi:10.1021/ol303231c |
| 56. | Bera, M. K.; Gholap, S. L.; Hommes, P.; Neuthe, K.; Trawny, D.; Rabe, J. P.; Lentz, D.; Zimmer, R.; Reissig, H.-U. Adv. Synth. Catal. 2013, 355, 3463–3474. doi:10.1002/adsc.201300613 |
| 57. | Podolan, G.; Hettmanczyk, L.; Hommes, P.; Sarkar, B.; Reissig, H.-U. Eur. J. Org. Chem. 2015, 7317–7323. doi:10.1002/ejoc.201501163 |
| 58. | Trawny, D.; Vandromme, L.; Rabe, J. P.; Reissig, H. U. Eur. J. Org. Chem. 2014, 4985–4992. doi:10.1002/ejoc.201402178 |
| 59. | Dash, J.; Trawny, D.; Rabe, J. P.; Reissig, H. U. Synlett 2015, 26, 1486–1489. doi:10.1055/s-0034-1380716 |
| 48. | Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220 |
| 49. | Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056 |
| 50. | Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787 |
| 51. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 52. | Bera, M. K.; Domínguez, M.; Hommes, P.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 394–404. doi:10.3762/bjoc.10.37 |
| 41. | Moriarty, R. M.; Vaid, R. K.; Ravikumar, V. T.; Vaid, B. K.; Hopkins, T. E. Tetrahedron 1988, 44, 1603–1607. doi:10.1016/S0040-4020(01)86720-X |
| 42. | Zheng, Y.; Li, X.; Ren, C.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. J. Org. Chem. 2012, 77, 10353–10361. doi:10.1021/jo302073e |
| 43. | Hempel, C.; Nachtsheim, B. J. Synthesis 2013, 24, 2119–2123. doi:10.1055/s-0033-1339491 |
| 44. | Zheng, M.; Huang, L.; Huang, H.; Li, X.; Wu, W.; Jiang, H. Org. Lett. 2014, 16, 5906–5909. doi:10.1021/ol502916a |
| 45. | Guo, W.-C.; Hu, F.; Huo, Y.-M.; Chang, H.-H.; Li, X.; Wie, W.-L. Org. Lett. 2015, 17, 3914–3917. doi:10.1021/acs.orglett.5b01933 |
| 46. | Lechel, T.; Lentz, D.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386 |
| 47. | Lechel, T.; Gerhard, M.; Trawny, D.; Brusilowskij, B.; Schefzig, L.; Zimmer, R.; Rabe, J. P.; Lentz, D.; Schalley, C. A.; Reissig, H.-U. Chem. – Eur. J. 2011, 17, 7480–7491. doi:10.1002/chem.201100382 |
© 2016 Hommes and Reissig; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)