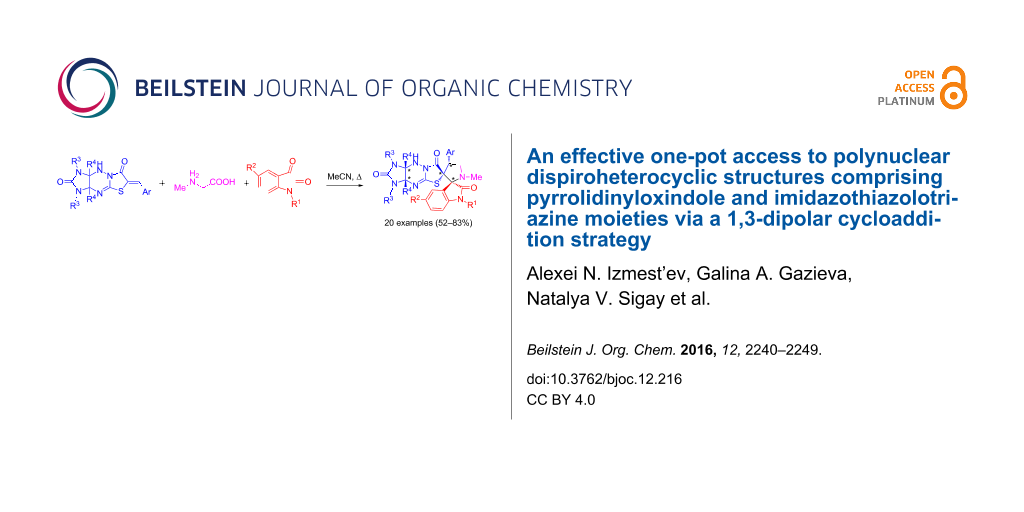
Abstract
An effective and highly regio- and diastereoselective one-pot method for the synthesis of new polynuclear dispiroheterocyclic systems with five stereogenic centers (dispiro[imidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine-6,3′-pyrrolidine-2′,3′′-indoles]) comprising pyrrolidinyloxindole and imidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine moieties has been developed. The method relies on a 1,3-dipolar cycloaddition of azomethine ylides generated in situ from isatin derivatives and sarcosine to 6-benzylideneimidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine-2,7-diones.

Graphical Abstract
Introduction
A global trend in modern organic chemistry is the design of molecular systems with various degrees of complexity to maximize the incorporation of useful properties while optimizing cost and efficiency [1]. A very extended and powerful approach for constructing complex N-heterocyclic systems in a regio and stereocontrolled fashion is the 1,3-dipolar cycloaddition of azomethine ylides to electron-deficient alkenes as dipolarophiles [2-8]. The in situ preparation of azomethine ylides from different carbonyl and amino components makes the cycloaddition one of the most valuable means of combinatorial chemistry. Such multicomponent reactions are characterized by productivity, operational simplicity, and efficiency [9-13]. A highly interesting class of heterocycles which is accessible through 1,3-dipolar cycloaddition reactions are compounds having the spiropyrrolidinyloxindole core [14-18]. The 3,3’-spiropyrrolidinyloxindole unit is found in the molecular skeleton of a large family of natural alkaloids with remarkable bioactivity profiles and interesting structural properties [19,20]. Derivatives with the 2,3’-spiropyrrolidinyloxindole core show various biological effects such as bactericidal and fungicidal [21], anticancer [22], cytotoxic to MCF-7 and HepG2 cells [23,24], and advanced glycation end (AGE) product formation inhibitory activities [25] (Figure 1).
![[1860-5397-12-216-1]](/bjoc/content/figures/1860-5397-12-216-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bioactive 2,3’-spiropyrrolidinyloxindoles.
Figure 1: Bioactive 2,3’-spiropyrrolidinyloxindoles.
Over the last decade, a lot of publications have been devoted to the synthesis of dispiro compounds comprising pyrrolidine, oxindole, and other heterocycle moieties and to the evaluation of their physiological properties [24,26-32]. In this regard, our attention was directed towards hetero-annelated 1,2,4-triazines, because this motif is part of many natural and synthetic bioactive products [33-40]. The pyrimido[5,4-e]-1,2,4-triazine constitutes the core of the antibiotics fervenulin, xanthothricin, and reumycin [33,34]. Other hetero-annelated 1,2,4-triazines reveal antiviral effect against influenza A and B viruses [35], anti-HIV and anticancer [36,37], antimicrobial and antifungal activities as well as cytotoxicity to MCF-7 cells [38,39]. Based on these observations we therefore aimed at combining the spiropyrrolidinyloxindole motif with hetero-annelated 1,2,4-triazine scaffolds.
Recently, we have already combined the imidazothiazolotriazine and 3,3’-spiropyrrolidinyloxindole moieties by a 1,3-dipolar cycloaddition of an azomethine ylide generated in situ from paraformaldehyde and sarcosine to oxoindolylidene derivatives of imidazothiazolotriazine. During this work we have found that the “small” azomethine ylide generated from paraformaldehyde and sarcosine approaches the double bond plane in (oxoindolylidene)imidazothiazolotriazines mainly from the side of the imidazolidine ring opposite to the phenyl groups (syn attack) (Scheme 1) [5].
![[1860-5397-12-216-i1]](/bjoc/content/inline/1860-5397-12-216-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Earlier studied cycloaddition reaction.
Scheme 1: Earlier studied cycloaddition reaction.
To further expand the spectrum of biological activity, it is of interest to synthesize the other types of spiro compounds such as the 2,3’-spiropyrrolidinyloxindole structure which is isosteric with the 3,3’-spiropyrrolidinyloxindole and to study the diastereoselectivity of its formation. It could be expected that the cycloaddition of more bulky azomethine ylides generated from isatins and sarcosine to benzylidene derivatives of the same imidazothiazolotriazine will proceed from the less sterically hindered side [41] (anti attack).
Herein, we report a regio- and diastereoselective one-pot method for the synthesis of a novel class of polynuclear dispiroheterocyclic structures comprising 2,3’-spiropyrrolidinyloxindole and imidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine moieties. The synthesis is based on a 1,3-dipolar cycloaddition of azomethine ylides generated in situ from isatins and sarcosine to tailor-made 6-benzylideneimidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine-2,7-diones.
Results and Discussion
The required dipolarophiles 1a–c were prepared by the three-component condensation of imidazotriazinethione 2, bromoacetic acid, and aromatic aldehydes (Scheme 2), as was described earlier by us [41]. The starting compound 2 is readily accessible and can be synthesized from 4,5-dihydroxy-4,5-diphenylimidazolidine-2-one [42] and thiosemicarbazide in 96% yield [43].
![[1860-5397-12-216-i2]](/bjoc/content/inline/1860-5397-12-216-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of dipolarophiles 1a–c.
Scheme 2: Synthesis of dipolarophiles 1a–c.
To optimize the 1,3-dipolar cycloaddition reaction conditions, 4-bromobenzylidene derivative 1a was chosen as a model substrate in the reaction with sarcosine and isatin 3a. The solvent, reaction time, and temperature were varied (Table 1).
Table 1: Optimization of the 1,3-dipolar cycloaddition reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-216-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Temp. (°C) | Time (h) | Yield (%)b |
|---|---|---|---|---|
| 1 | EtOH | reflux | 24 | 20 |
| 2 | EtOH | reflux | 42 | 23 |
| 3 | toluene | reflux | 24 | 15 |
| 4 | CHCl3 | reflux | 24 | 50 |
| 5 | CHCl3 | reflux | 42 | 59 |
| 6 | MeCN | reflux | 24 | 56 |
| 7 | MeCN | reflux | 30 | 71 |
| 8 | MeCN | reflux | 36 | 78 |
| 9 | MeCN | reflux | 42 | 79 |
| 10 | MeCN | rt | 42 | 0 |
aReaction conditions: heating the mixture of compound 1a (0.5 mmol), isatin (3a, 0.5 mmol), and sarcosine (0.5 mmol) in the corresponding solvent (40 mL) for the indicated time. bIsolated yield.
As can be seen from Table 1, the cycloaddition was carried out in different solvents such as ethanol, chloroform, acetonitrile, and toluene. When the reaction was performed in ethanol or toluene, product 4a was obtained in only low yields (Table 1, entries 1–3). Slightly increased yields were achieved in refluxing chloroform (Table 1, entries 4 and 5) and the best results were obtained in refluxing acetonitrile (Table 1, entries 6–9). At room temperature, the reaction does not proceed at all (Table 1, entry 10). Changing the reaction time from 24 h to 30 h significantly improved the yield of product 4a whereas a further increase of the reaction time (>36 h) did not further improve the yield (Table 1, entries 6–9). The reaction progress was monitored by recording 1H NMR spectra of samples taken from the reaction mixture after 24, 30, 36, and 42 h. The formation of the pyrrolidine ring was detected by the appearance of triplets for the ring CH2 and CH group protons at 3.56, 3.95, and 4.45 ppm. Doublets for the C-3a and C-9a phenyl ortho-protons in compound 4a (6.13 and 6.55 ppm, respectively) were observed at higher field than those of compound 1a (6.75 and 6.83 ppm). The integrated intensity ratio of these protons was used to calculate the ratio of the target and starting compounds.
With the optimized conditions in hand, we next investigated the substrate scope for the reaction. First, various isatins 3a–e were used for the generation of the azomethine ylides for the cycloaddition with compound 1a (Scheme 3, Figure 2).
![[1860-5397-12-216-i3]](/bjoc/content/inline/1860-5397-12-216-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of dispirocompounds 4a–o.
Scheme 3: Synthesis of dispirocompounds 4a–o.
![[1860-5397-12-216-2]](/bjoc/content/figures/1860-5397-12-216-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesis of dispiro compounds 4a–o. Reaction conditions: heating the mixture of compounds 1 (0.5 mmol), isatins 3 (0.5 mmol), and sarcosine (0.5 mmol) in acetonitrile (40 mL) for 36 h.
Figure 2: Synthesis of dispiro compounds 4a–o. Reaction conditions: heating the mixture of compounds 1 (0.5 m...
It was found that apart from model substrate 3a, N-alkyl- (3b,c), N-allyl- (3d) and N-methyl-5-bromoisatins (3e) reacted with sarcosine and dipolarophile 1a to afford the desired products 4a–e in 54–78% yields. Next, the nitrobenzylidene and benzylidene derivatives 1b,c were subjected to the reaction with isatins 3a–e and sarcosine under the optimized conditions to afford the dispiro compounds 4f–o in 52–83% yields. As shown in Figure 2, the best yields of the cycloadducts 4 were observed in the reaction of unsubstituted isatin (3a) as the carbonyl component for the generation of the azomethine ylide as well as for nitrobenzylidene derivative 1b as dipolarophile.
To further extend the substrate scope of this reaction, we used benzylidene derivatives of other imidazothiazolotriazines 1d–f without substituents at the bridge carbon atoms C(3a) and C(9a). The previously unknown compounds 1d–f were synthesized in good yields by the condensation of imidazothiazolotriazines 5a,b [44] with the corresponding aromatic aldehydes (Scheme 4).
![[1860-5397-12-216-i4]](/bjoc/content/inline/1860-5397-12-216-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of dipolarophiles 1d–f.
Scheme 4: Synthesis of dipolarophiles 1d–f.
These derivatives cannot be prepared by a three-component condensation of imidazotriazinethione with bromoacetic acid and an aromatic aldehyde, similarly to the synthesis of compounds 1a–c. The reaction of aromatic aldehydes with imidazotriazinethiones without phenyl substituents in acidic media results in hydrazone formation and triazine-ring contraction [45].
The reaction of compounds 1d–f with sarcosine and isatins 3a,d,f also proceeded successfully, but required the addition of chloroform to the reaction mixture and an increased reaction time of 72 h. The novel dispiro compounds 4p–t were finally obtained in 55–74% yields (Figure 3).
![[1860-5397-12-216-3]](/bjoc/content/figures/1860-5397-12-216-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Synthesis of dispiro compounds 4p–t. Reaction conditions: heating the solution of compounds 1 (0.5 mmol), isatins 3 (0.5 mmol), and sarcosine (0.5 mmol) in the mixture of acetonitrile (30 mL) and chloroform (10 mL) for 72 h.
Figure 3: Synthesis of dispiro compounds 4p–t. Reaction conditions: heating the solution of compounds 1 (0.5 ...
The structures of the synthesized compounds as well as the regioselectivity and diastereoselectivity of the cycloaddition were elucidated by spectroscopic methods and single crystal X-ray diffraction. The cycloadducts 4a–t were characterized by IR, NMR, and HRMS analytical methods. The IR spectra of compounds 4 showed three intense absorption bands at 1728–1697, 1709–1680 (in some cases instead of these two bands, one broad band is observed), and 1649–1634 cm−1 that are characteristic of oxindole, imidazolidinone, and thiazolidinone ring carbonyl groups. The 1H NMR spectra of compounds 4 exhibited, along with the proton signals of the imidazothiazolotriazine and oxindole moieties, the signals for pyrrolidine ring protons: a singlet at 2.05–2.18 ppm for N(1’)Me group protons, two triplets at 3.49–3.66 and 3.93–4.05 ppm assignable to the protons of methylene C(5’)H2 group, and one triplet at 4.45–4.70 ppm corresponding to the proton of C(4’)H. This clearly demonstrates the regiochemistry of the cycloaddition. If the other possible regioisomer had formed, the 1H NMR spectra would have shown a singlet for the C(4’)H proton.
In more detail, the structure of compounds 4 was studied on the example of 4f by COSY, {1H-13C}HSQC, {1H-13C}- and {1H-15N}HMBC NMR experiments. For instance, in the {1H-13C}HMBC spectrum of 4f, the N(1’)Me protons (2.17 ppm) correlate with the C-5’ (57.52 ppm) and spiro C-2’ (80.29 ppm) carbons; the proton of C(4’)H (4.63 ppm) of the pyrrolidine ring correlates with the spiro carbon C-3’ (68.47 ppm) and the carbon atoms of the C(5’)H2 and C(7)=O (167.31 ppm) groups (Figure 4). The correlations of the C(5’)H2 group protons are different. One of them (4.02 ppm) shows cross-peaks with a neighboring C-4’ carbon atom (50.90 ppm) and the carbon atom of the N(1’)Me (35.38 ppm) group. Another one (3.61 ppm), correlates with both spiro carbon atoms C-2’ and C-3’ (see Supporting Information File 1 for full experimental data).
![[1860-5397-12-216-4]](/bjoc/content/figures/1860-5397-12-216-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Key interactions in {1H-13C}HMBC spectrum of 4f.
Figure 4: Key interactions in {1H-13C}HMBC spectrum of 4f.
Finally, the regio- and stereochemistry of the cycloaddition were confirmed by single crystal X-ray diffraction analysis of compounds 4c (Figure 5), 4e (Figure 6), and 4r (Figure 7) (see Supporting Information Files 2–4). The relative configurations of the stereocenters of compounds 4 are 2’R*, 3aS*, 3’R*, 4’R*, 9aR*.
![[1860-5397-12-216-5]](/bjoc/content/figures/1860-5397-12-216-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: General view of 4c in the crystal in thermal ellipsoids representation (50% probability). Hydrogen atoms connected to carbon atoms are omitted for clarity.
Figure 5: General view of 4c in the crystal in thermal ellipsoids representation (50% probability). Hydrogen ...
![[1860-5397-12-216-6]](/bjoc/content/figures/1860-5397-12-216-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: General view of 4e in the crystal in thermal ellipsoids representation (40% probability). Hydrogen atoms connected to carbon atoms are omitted for clarity.
Figure 6: General view of 4e in the crystal in thermal ellipsoids representation (40% probability). Hydrogen ...
![[1860-5397-12-216-7]](/bjoc/content/figures/1860-5397-12-216-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: General view of 4r in the crystal in thermal ellipsoids representation (50% probability). Hydrogen atoms connected to carbon atoms are omitted for clarity.
Figure 7: General view of 4r in the crystal in thermal ellipsoids representation (50% probability). Hydrogen ...
The homogeneity of compounds 4b–d,f–i,l–n was additionally confirmed by powder X-ray diffraction. The analysis of the experimental powder diffraction patterns of compounds 4b–d,f–i,l–n show that the investigated samples were single phase (see Supporting Information File 1). Thus, the cycloaddition of azomethine ylides was found to be highly regioselective, as the electron-rich carbon of the dipole adds to the β-carbon of the α,β-unsaturated moiety of dipolarophile 1. Further the reaction is diastereoselective, as only one diastereomer is obtained in good to high yields, although multiple (five) stereocenters are present in products 4.
The possible approaches of the azomethine ylide are shown in Figure 8. The X-ray diffraction structures of 4c, 4e, and 4r reflect that the cycloaddition proceeds via an exo-transition state, because the corresponding endo-transition state would require more energy of activation, as it would result in an electrostatic repulsion between the cis carbonyls thus increasing the free energy of activation [46,47]. As expected, the azomethine ylide adds at the double bond of 1a–c from that side in which the phenyl substituents are directed (anti-exo).
![[1860-5397-12-216-8]](/bjoc/content/figures/1860-5397-12-216-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Modes of approach of azomethine ylide (R = H, Ph).
Figure 8: Modes of approach of azomethine ylide (R = H, Ph).
Conclusion
In summary, a simple, general, and efficient one-pot method for the construction of previously unknown substituted dispiro[imidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine-6,3′-pyrrolidine-2′,3′′-indoles] was developed. This method is based on the highly regio- and diastereoselective 1,3-dipolar cycloaddition reaction of azomethine ylides generated in situ from various isatin derivatives and sarcosine to readily available 6-benzylideneimidazo[4,5-e]thiazolo[3,2-b]-1,2,4-triazine-2,7-diones. The synthesized structures represent a new class of promising bioactive polynuclear dispiroheterocyclic structures comprising pyrrolidinyloxindole and imidazothiazolotriazine moieties. Investigations of cytotoxic activities of the synthesized products against A549, HCT116, RD, and MCF7 cell lines are in progress.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 4.5 MB | Download |
| Supporting Information File 2: CIF file for compound 4c. | ||
| Format: CIF | Size: 2.3 MB | Download |
| Supporting Information File 3: CIF file for compound 4e. | ||
| Format: CIF | Size: 4.7 MB | Download |
| Supporting Information File 4: CIF file for compound 4r. | ||
| Format: CIF | Size: 1.2 MB | Download |
References
-
Ananikov, V. P.; Khemchyan, L. L.; Ivanova, Yu. V.; Bukhtiyarov, V. I.; Sorokin, A. M.; Prosvirin, I. P.; Vatsadze, S. Z.; Medved'ko, A. V.; Nuriev, V. N.; Dilman, A. D.; Levin, V. V.; Koptyug, I. V.; Kovtunov, K. V.; Zhivonitko, V. V.; Likholobov, V. A.; Romanenko, A. V.; Simonov, P. A.; Nenajdenko, V. G.; Shmatova, O. I.; Muzalevskiy, V. M.; Nechaev, M. S.; Asachenko, A. F.; Morozov, O. S.; Dzhevakov, P. B.; Osipov, S. N.; Vorobyeva, D. V.; Topchiy, M. A.; Zotova, M. A.; Ponomarenko, S. A.; Borshchev, O. V.; Luponosov, Yu. N.; Rempel, A. A.; Valeeva, A. A.; Stakheev, A. Yu.; Turova, O. V.; Mashkovsky, I. S.; Sysolyatin, S. V.; Malykhin, V. V.; Bukhtiyarova, G. A.; Terent'ev, A. O.; Krylov, I. B. Russ. Chem. Rev. 2014, 83, 885–985. doi:10.1070/RC2014v83n10ABEH004471
Return to citation in text: [1] -
Narayan, R.; Potowski, M.; Jia, Z.-J.; Antonchick, A. P.; Waldmann, H. Acc. Chem. Res. 2014, 47, 1296–1310. doi:10.1021/ar400286b
Return to citation in text: [1] -
Borad, M. A.; Bhoi, M. N.; Prajapati, N. P.; Patel, H. D. Synth. Commun. 2014, 44, 1043–1057. doi:10.1080/00397911.2013.858361
Return to citation in text: [1] -
Kissane, M.; Maguire, A. R. Chem. Soc. Rev. 2010, 39, 845–883. doi:10.1039/B909358N
Return to citation in text: [1] -
Gazieva, G. A.; Kolotyrkina, N. G.; Kravchenko, A. N.; Makhova, N. N. Russ. Chem. Bull. 2014, 63, 431–434. doi:10.1007/s11172-014-0449-2
Return to citation in text: [1] [2] -
Sosnovskikh, V. Y.; Kornev, M. Y.; Moshkin, V. S.; Buev, E. M. Tetrahedron 2014, 70, 9253–9261. doi:10.1016/j.tet.2014.09.090
Return to citation in text: [1] -
Najera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594
Return to citation in text: [1] -
Bharitkar, Y. P.; Kanhar, S.; Suneel, N.; Mondal, S. K.; Hazra, A.; Mondal, N. B. Mol. Diversity 2015, 19, 251–261. doi:10.1007/s11030-015-9574-6
Return to citation in text: [1] -
Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, 2006. doi:10.1002/9783527609925
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180
Return to citation in text: [1] -
Borad, M. A.; Bhoi, M. N.; Prajapati, N. P.; Patel, H. D. Synth. Commun. 2014, 44, 897–922. doi:10.1080/00397911.2013.843196
Return to citation in text: [1] -
Sun, J.; Chen, L.; Gong, H.; Yan, C.-G. Org. Biomol. Chem. 2015, 13, 5905–5917. doi:10.1039/C5OB00437C
Return to citation in text: [1] -
Karnakar, K.; Ramesh, K.; Reddy, K. H. V.; Anil Kumar, B. S. P.; Nanubonula, J. B.; Nageswar, Y. V. D. New J. Chem. 2015, 39, 8978–8983. doi:10.1039/C5NJ01448D
Return to citation in text: [1] -
Xia, M.; Ma, R.-Z. J. Heterocycl. Chem. 2014, 51, 539–554. doi:10.1002/jhet.1114
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735–740. doi:10.1038/nchem.730
Return to citation in text: [1] -
Wang, C.-S.; Zhu, R.-Y.; Zheng, J.; Shi, F.; Tu, S.-J. J. Org. Chem. 2015, 80, 512–520. doi:10.1021/jo502516e
Return to citation in text: [1] -
Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8
Return to citation in text: [1] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Pardasani, P.; Pardasani, R. T.; Chaturvedi, V.; Saxena, A. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2003, 42B, 412–415.
Return to citation in text: [1] -
Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] -
Tan, W.; Zhu, X.-T.; Zhang, S.; Xing, G.-J.; Zhu, R.-Y.; Shi, F. RSC Adv. 2013, 3, 10875–10886. doi:10.1039/c3ra40874d
Return to citation in text: [1] -
Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. doi:10.1016/j.bmcl.2015.06.076
Return to citation in text: [1] [2] -
Kaur, A.; Singh, B.; Vyas, B.; Silakari, O. Eur. J. Med. Chem. 2014, 79, 282–289. doi:10.1016/j.ejmech.2014.04.022
Return to citation in text: [1] -
Poornachandran, M.; Raghunathan, R. Tetrahedron 2006, 62, 11274–11281. doi:10.1016/j.tet.2006.09.008
Return to citation in text: [1] -
Karthikeyan, K.; Sivakumar, P. M.; Doble, M.; Perumal, P. T. Eur. J. Med. Chem. 2010, 45, 3446–3452. doi:10.1016/j.ejmech.2010.04.035
Return to citation in text: [1] -
Li, X.; Liu, B.; Liu, H.; Yu, X.; Yi, P. J. Heterocycl. Chem. 2012, 49, 1050–1053. doi:10.1002/jhet.925
Return to citation in text: [1] -
Chandraprakash, K.; Sankaran, M.; Uvarani, C.; Shankar, R.; Ata, A.; Dallemer, F.; Mohan, P. S. Tetrahedron Lett. 2013, 54, 3896–3901. doi:10.1016/j.tetlet.2013.05.077
Return to citation in text: [1] -
Ivanenkov, Y. A.; Vasilevski, S. V.; Beloglazkina, E. K.; Kukushkin, M. E.; Machulkin, A. E.; Veselov, M. S.; Chufarova, N. V.; Chernyaginab, E. S.; Vanzcool, A. S.; Zyk, N. V.; Skvortsov, D. A.; Khutornenko, A. A.; Rusanov, A. L.; Tonevitsky, A. G.; Dontsova, O. A.; Majouga, A. G. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. doi:10.1016/j.bmcl.2014.09.070
Return to citation in text: [1] -
Anis'kov, A. A.; Kamneva, I. Ye.; Zheleznova, M. A.; Yegorova, A. Yu. Chem. Heterocycl. Compd. 2015, 51, 709–712. doi:10.1007/s10593-015-1763-9
Return to citation in text: [1] -
Kurbatov, S. V.; Zarubaev, V. V.; Karpinskaya, L. A.; Shvets, A. A.; Kletsky, M. E.; Burov, O. N.; Morozov, P. G.; Kiselev, O. I.; Minkin, V. I. Russ. Chem. Bull. 2014, 63, 1130–1136. doi:10.1007/s11172-014-0560-4
Return to citation in text: [1] -
Ruanpanun, P.; Laatsch, H.; Tangchitsomkid, N.; Lumyong, S. World J. Microbiol. Biotechnol. 2011, 27, 1373–1380. doi:10.1007/s11274-010-0588-z
Return to citation in text: [1] [2] -
Nagamatsu, T.; Yamasaki, H.; Hirota, T.; Yamato, M.; Kido, Y.; Shibata, M.; Yoneda, F. Chem. Pharm. Bull. 1993, 41, 362–368. doi:10.1248/cpb.41.362
Return to citation in text: [1] [2] -
Kiselev, O. I.; Deyeva, E. G.; Melnicova, T. I.; Kozeletskaia, K. N.; Kiselev, А. S.; Rusinov, V. L.; Charushin, V. N.; Chupahin, О. N. Vopr. Virusol. 2012, 57, 9–12.
Return to citation in text: [1] [2] -
Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Pharmazie 1994, 49, 729–733.
Return to citation in text: [1] [2] -
Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Boll. Chim. Farm. 1994, 133, 381–388.
Return to citation in text: [1] [2] -
Abd El-Moneim, M.; Hasanen, J. A.; El-Deen, I. M.; Abd El-Fattah, W. Res. Chem. Intermed. 2015, 41, 3543–3561. doi:10.1007/s11164-013-1470-z
Return to citation in text: [1] [2] -
El-Nassan, H. B. Eur. J. Med. Chem. 2012, 53, 22–27. doi:10.1016/j.ejmech.2012.03.028
Return to citation in text: [1] [2] -
Abraham, S.; Hadd, M. J.; Tran, L.; Vickers, T.; Sindac, J.; Milanov, Z. V.; Holladay, M. W.; Bhagwat, S. S.; Hua, H.; Ford Pulido, J. M.; Cramer, M. D.; Gitnick, D.; James, J.; Dao, A.; Belli, B.; Armstrong, R. C.; Treiber, D. K.; Liu, G. Bioorg. Med. Chem. Lett. 2011, 21, 5296–5300. doi:10.1016/j.bmcl.2011.07.027
Return to citation in text: [1] -
Gazieva, G. A.; Serkov, S. A.; Sigai, N. V.; Kostikova, N. N.; Nelyubina, Yu. V.; Shishkova, E. A.; Kravchenko, A. N. Chem. Heterocycl. Compd. 2013, 49, 1097–1101. doi:10.1007/s10593-013-1349-3
Return to citation in text: [1] [2] -
Neville, R. J. Org. Chem. 1958, 23, 1588–1590. doi:10.1021/jo01104a630
Return to citation in text: [1] -
Sigachev, A. S.; Kravchenko, A. N.; Lyssenko, K. A.; Belyakov, P. A.; Lebedev, O. V.; Makhova, N. N. Mendeleev Commun. 2003, 13, 190–191. doi:10.1070/MC2003v013n04ABEH001810
Return to citation in text: [1] -
Gazieva, G. A.; Poluboyarov, P. A.; Nelyubina, Yu. V.; Struchkova, M. I.; Kravchenko, A. N. Chem. Heterocycl. Compd. 2012, 48, 1382–1389. doi:10.1007/s10593-012-1147-3
Return to citation in text: [1] -
Gazieva, G. A.; Poluboyarov, P. A.; Popov, L. D.; Kolotyrkina, N. G.; Kravchenko, A. N.; Makhova, N. N. Synthesis 2012, 44, 3366–3370. doi:10.1055/s-0032-1317194
Return to citation in text: [1] -
He, J.; Ouyang, G.; Yuan, Z.; Tong, R.; Shi, J.; Ouyang, L. Molecules 2013, 18, 5142–5154. doi:10.3390/molecules18055142
Return to citation in text: [1] -
Kumar, R. R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. Tetrahedron 2008, 64, 2962–2971. doi:10.1016/j.tet.2008.01.072
Return to citation in text: [1]
| 41. | Gazieva, G. A.; Serkov, S. A.; Sigai, N. V.; Kostikova, N. N.; Nelyubina, Yu. V.; Shishkova, E. A.; Kravchenko, A. N. Chem. Heterocycl. Compd. 2013, 49, 1097–1101. doi:10.1007/s10593-013-1349-3 |
| 41. | Gazieva, G. A.; Serkov, S. A.; Sigai, N. V.; Kostikova, N. N.; Nelyubina, Yu. V.; Shishkova, E. A.; Kravchenko, A. N. Chem. Heterocycl. Compd. 2013, 49, 1097–1101. doi:10.1007/s10593-013-1349-3 |
| 1. | Ananikov, V. P.; Khemchyan, L. L.; Ivanova, Yu. V.; Bukhtiyarov, V. I.; Sorokin, A. M.; Prosvirin, I. P.; Vatsadze, S. Z.; Medved'ko, A. V.; Nuriev, V. N.; Dilman, A. D.; Levin, V. V.; Koptyug, I. V.; Kovtunov, K. V.; Zhivonitko, V. V.; Likholobov, V. A.; Romanenko, A. V.; Simonov, P. A.; Nenajdenko, V. G.; Shmatova, O. I.; Muzalevskiy, V. M.; Nechaev, M. S.; Asachenko, A. F.; Morozov, O. S.; Dzhevakov, P. B.; Osipov, S. N.; Vorobyeva, D. V.; Topchiy, M. A.; Zotova, M. A.; Ponomarenko, S. A.; Borshchev, O. V.; Luponosov, Yu. N.; Rempel, A. A.; Valeeva, A. A.; Stakheev, A. Yu.; Turova, O. V.; Mashkovsky, I. S.; Sysolyatin, S. V.; Malykhin, V. V.; Bukhtiyarova, G. A.; Terent'ev, A. O.; Krylov, I. B. Russ. Chem. Rev. 2014, 83, 885–985. doi:10.1070/RC2014v83n10ABEH004471 |
| 19. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050 |
| 20. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 38. | Abd El-Moneim, M.; Hasanen, J. A.; El-Deen, I. M.; Abd El-Fattah, W. Res. Chem. Intermed. 2015, 41, 3543–3561. doi:10.1007/s11164-013-1470-z |
| 39. | El-Nassan, H. B. Eur. J. Med. Chem. 2012, 53, 22–27. doi:10.1016/j.ejmech.2012.03.028 |
| 14. | Xia, M.; Ma, R.-Z. J. Heterocycl. Chem. 2014, 51, 539–554. doi:10.1002/jhet.1114 |
| 15. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 16. | Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735–740. doi:10.1038/nchem.730 |
| 17. | Wang, C.-S.; Zhu, R.-Y.; Zheng, J.; Shi, F.; Tu, S.-J. J. Org. Chem. 2015, 80, 512–520. doi:10.1021/jo502516e |
| 18. | Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8 |
| 5. | Gazieva, G. A.; Kolotyrkina, N. G.; Kravchenko, A. N.; Makhova, N. N. Russ. Chem. Bull. 2014, 63, 431–434. doi:10.1007/s11172-014-0449-2 |
| 9. | Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, 2006. doi:10.1002/9783527609925 |
| 10. | Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180 |
| 11. | Borad, M. A.; Bhoi, M. N.; Prajapati, N. P.; Patel, H. D. Synth. Commun. 2014, 44, 897–922. doi:10.1080/00397911.2013.843196 |
| 12. | Sun, J.; Chen, L.; Gong, H.; Yan, C.-G. Org. Biomol. Chem. 2015, 13, 5905–5917. doi:10.1039/C5OB00437C |
| 13. | Karnakar, K.; Ramesh, K.; Reddy, K. H. V.; Anil Kumar, B. S. P.; Nanubonula, J. B.; Nageswar, Y. V. D. New J. Chem. 2015, 39, 8978–8983. doi:10.1039/C5NJ01448D |
| 35. | Kiselev, O. I.; Deyeva, E. G.; Melnicova, T. I.; Kozeletskaia, K. N.; Kiselev, А. S.; Rusinov, V. L.; Charushin, V. N.; Chupahin, О. N. Vopr. Virusol. 2012, 57, 9–12. |
| 2. | Narayan, R.; Potowski, M.; Jia, Z.-J.; Antonchick, A. P.; Waldmann, H. Acc. Chem. Res. 2014, 47, 1296–1310. doi:10.1021/ar400286b |
| 3. | Borad, M. A.; Bhoi, M. N.; Prajapati, N. P.; Patel, H. D. Synth. Commun. 2014, 44, 1043–1057. doi:10.1080/00397911.2013.858361 |
| 4. | Kissane, M.; Maguire, A. R. Chem. Soc. Rev. 2010, 39, 845–883. doi:10.1039/B909358N |
| 5. | Gazieva, G. A.; Kolotyrkina, N. G.; Kravchenko, A. N.; Makhova, N. N. Russ. Chem. Bull. 2014, 63, 431–434. doi:10.1007/s11172-014-0449-2 |
| 6. | Sosnovskikh, V. Y.; Kornev, M. Y.; Moshkin, V. S.; Buev, E. M. Tetrahedron 2014, 70, 9253–9261. doi:10.1016/j.tet.2014.09.090 |
| 7. | Najera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594 |
| 8. | Bharitkar, Y. P.; Kanhar, S.; Suneel, N.; Mondal, S. K.; Hazra, A.; Mondal, N. B. Mol. Diversity 2015, 19, 251–261. doi:10.1007/s11030-015-9574-6 |
| 36. | Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Pharmazie 1994, 49, 729–733. |
| 37. | Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Boll. Chim. Farm. 1994, 133, 381–388. |
| 25. | Kaur, A.; Singh, B.; Vyas, B.; Silakari, O. Eur. J. Med. Chem. 2014, 79, 282–289. doi:10.1016/j.ejmech.2014.04.022 |
| 33. | Ruanpanun, P.; Laatsch, H.; Tangchitsomkid, N.; Lumyong, S. World J. Microbiol. Biotechnol. 2011, 27, 1373–1380. doi:10.1007/s11274-010-0588-z |
| 34. | Nagamatsu, T.; Yamasaki, H.; Hirota, T.; Yamato, M.; Kido, Y.; Shibata, M.; Yoneda, F. Chem. Pharm. Bull. 1993, 41, 362–368. doi:10.1248/cpb.41.362 |
| 35. | Kiselev, O. I.; Deyeva, E. G.; Melnicova, T. I.; Kozeletskaia, K. N.; Kiselev, А. S.; Rusinov, V. L.; Charushin, V. N.; Chupahin, О. N. Vopr. Virusol. 2012, 57, 9–12. |
| 36. | Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Pharmazie 1994, 49, 729–733. |
| 37. | Abdel-Rahman, R. M.; Seada, M.; Fawzy, M.; El-Baz, I. Boll. Chim. Farm. 1994, 133, 381–388. |
| 38. | Abd El-Moneim, M.; Hasanen, J. A.; El-Deen, I. M.; Abd El-Fattah, W. Res. Chem. Intermed. 2015, 41, 3543–3561. doi:10.1007/s11164-013-1470-z |
| 39. | El-Nassan, H. B. Eur. J. Med. Chem. 2012, 53, 22–27. doi:10.1016/j.ejmech.2012.03.028 |
| 40. | Abraham, S.; Hadd, M. J.; Tran, L.; Vickers, T.; Sindac, J.; Milanov, Z. V.; Holladay, M. W.; Bhagwat, S. S.; Hua, H.; Ford Pulido, J. M.; Cramer, M. D.; Gitnick, D.; James, J.; Dao, A.; Belli, B.; Armstrong, R. C.; Treiber, D. K.; Liu, G. Bioorg. Med. Chem. Lett. 2011, 21, 5296–5300. doi:10.1016/j.bmcl.2011.07.027 |
| 45. | Gazieva, G. A.; Poluboyarov, P. A.; Popov, L. D.; Kolotyrkina, N. G.; Kravchenko, A. N.; Makhova, N. N. Synthesis 2012, 44, 3366–3370. doi:10.1055/s-0032-1317194 |
| 23. | Tan, W.; Zhu, X.-T.; Zhang, S.; Xing, G.-J.; Zhu, R.-Y.; Shi, F. RSC Adv. 2013, 3, 10875–10886. doi:10.1039/c3ra40874d |
| 24. | Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. doi:10.1016/j.bmcl.2015.06.076 |
| 33. | Ruanpanun, P.; Laatsch, H.; Tangchitsomkid, N.; Lumyong, S. World J. Microbiol. Biotechnol. 2011, 27, 1373–1380. doi:10.1007/s11274-010-0588-z |
| 34. | Nagamatsu, T.; Yamasaki, H.; Hirota, T.; Yamato, M.; Kido, Y.; Shibata, M.; Yoneda, F. Chem. Pharm. Bull. 1993, 41, 362–368. doi:10.1248/cpb.41.362 |
| 46. | He, J.; Ouyang, G.; Yuan, Z.; Tong, R.; Shi, J.; Ouyang, L. Molecules 2013, 18, 5142–5154. doi:10.3390/molecules18055142 |
| 47. | Kumar, R. R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. Tetrahedron 2008, 64, 2962–2971. doi:10.1016/j.tet.2008.01.072 |
| 22. | Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056 |
| 43. | Sigachev, A. S.; Kravchenko, A. N.; Lyssenko, K. A.; Belyakov, P. A.; Lebedev, O. V.; Makhova, N. N. Mendeleev Commun. 2003, 13, 190–191. doi:10.1070/MC2003v013n04ABEH001810 |
| 21. | Pardasani, P.; Pardasani, R. T.; Chaturvedi, V.; Saxena, A. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2003, 42B, 412–415. |
| 24. | Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. doi:10.1016/j.bmcl.2015.06.076 |
| 26. | Poornachandran, M.; Raghunathan, R. Tetrahedron 2006, 62, 11274–11281. doi:10.1016/j.tet.2006.09.008 |
| 27. | Karthikeyan, K.; Sivakumar, P. M.; Doble, M.; Perumal, P. T. Eur. J. Med. Chem. 2010, 45, 3446–3452. doi:10.1016/j.ejmech.2010.04.035 |
| 28. | Li, X.; Liu, B.; Liu, H.; Yu, X.; Yi, P. J. Heterocycl. Chem. 2012, 49, 1050–1053. doi:10.1002/jhet.925 |
| 29. | Chandraprakash, K.; Sankaran, M.; Uvarani, C.; Shankar, R.; Ata, A.; Dallemer, F.; Mohan, P. S. Tetrahedron Lett. 2013, 54, 3896–3901. doi:10.1016/j.tetlet.2013.05.077 |
| 30. | Ivanenkov, Y. A.; Vasilevski, S. V.; Beloglazkina, E. K.; Kukushkin, M. E.; Machulkin, A. E.; Veselov, M. S.; Chufarova, N. V.; Chernyaginab, E. S.; Vanzcool, A. S.; Zyk, N. V.; Skvortsov, D. A.; Khutornenko, A. A.; Rusanov, A. L.; Tonevitsky, A. G.; Dontsova, O. A.; Majouga, A. G. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. doi:10.1016/j.bmcl.2014.09.070 |
| 31. | Anis'kov, A. A.; Kamneva, I. Ye.; Zheleznova, M. A.; Yegorova, A. Yu. Chem. Heterocycl. Compd. 2015, 51, 709–712. doi:10.1007/s10593-015-1763-9 |
| 32. | Kurbatov, S. V.; Zarubaev, V. V.; Karpinskaya, L. A.; Shvets, A. A.; Kletsky, M. E.; Burov, O. N.; Morozov, P. G.; Kiselev, O. I.; Minkin, V. I. Russ. Chem. Bull. 2014, 63, 1130–1136. doi:10.1007/s11172-014-0560-4 |
| 44. | Gazieva, G. A.; Poluboyarov, P. A.; Nelyubina, Yu. V.; Struchkova, M. I.; Kravchenko, A. N. Chem. Heterocycl. Compd. 2012, 48, 1382–1389. doi:10.1007/s10593-012-1147-3 |
© 2016 Izmest’ev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)