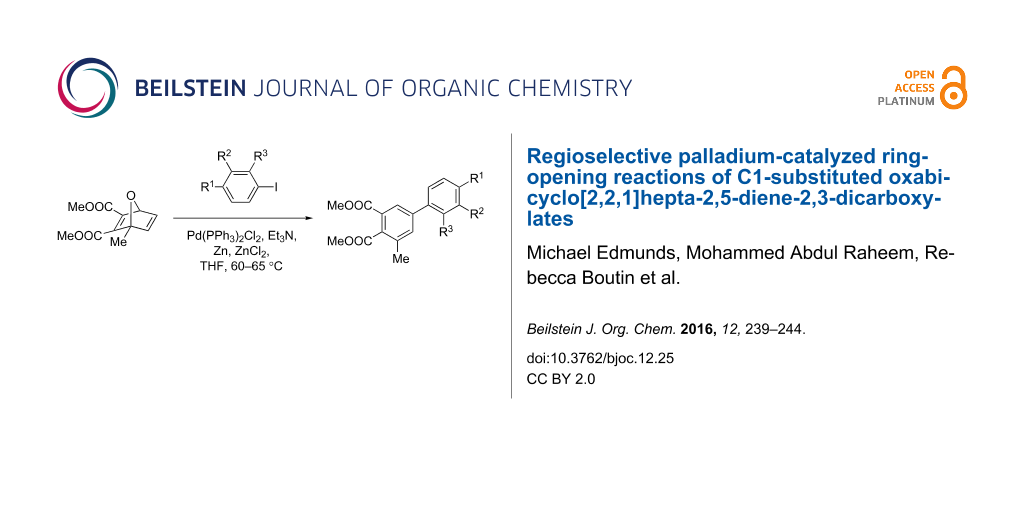
Abstract
Palladium-catalyzed ring-opening reactions of C1 substituted 7-oxanorbornadiene derivatives with aryl iodides were investigated. The optimal conditions for this reaction were found to be PdCl2(PPh3)2, ZnCl2, Et3N and Zn in THF. Both steric and electronic factors played a role in the outcome of the reaction as increasing the steric bulk on the bridgehead carbon decreased the yield. These reactions were found to be highly regioselective, giving only one of the two possible regioisomers in all cases. A diverse collection of novel, highly substituted biphenyl derivatives were obtained.

Graphical Abstract
Introduction
Palladium-catalyzed ring-opening reactions of 7-oxanorbornadiene with aryl iodides produce unsymmetrical, highly-substituted biphenyl derivatives making this reaction a very useful tool in organic synthesis [1-6]. The products of these reactions are also phthalates which have been used as intermediates in the synthesis of pharmaceuticals and biologically active agents, such as the anthracyclinone daunomycinone [7]. Phthalates are also used in paints, cosmetics, and as plasticizers in polymeric materials [8].
Although there are several examples of palladium-catalyzed ring-opening reactions of oxabenzonorbornadiene (Scheme 1), there is relatively little literature regarding reactions of non-aromatic 7-oxanorbornadiene systems [9-13]. To our knowledge, there has only been one example of 1 undergoing a ring-opening reaction, which used p-iodotoluene and a palladium catalyst (Scheme 2) [14]. The result was the addition of the aryl group to the unsubstituted double bond followed by dehydration to give an unsymmetrical biphenyl derivative. However, there have not been any investigations into the effect C1 substitution of 1 would have on the reaction. The addition of a substituent at the bridgehead carbon renders the bicyclic structure unsymmetrical and alters the steric and electronic factors imposed on the incoming palladium–aryl complex, which could affect the reactivity and regioselectivity of the reaction. Two regioisomers are possible for this reaction based on whether the aryl group is added to carbon a or carbon b of 2 (Scheme 3).
![[1860-5397-12-25-i1]](/bjoc/content/inline/1860-5397-12-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Palladium-catalyzed ring-opening reactions of oxabenzonorbornadiene.
Scheme 1: Palladium-catalyzed ring-opening reactions of oxabenzonorbornadiene.
![[1860-5397-12-25-i2]](/bjoc/content/inline/1860-5397-12-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Palladium-catalyzed ring-opening of 1 with p-iodotoluene.
Scheme 2: Palladium-catalyzed ring-opening of 1 with p-iodotoluene.
![[1860-5397-12-25-i3]](/bjoc/content/inline/1860-5397-12-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Potential regioisomers from the palladium-catalyzed ring-opening reaction of 2 with aryl iodides.
Scheme 3: Potential regioisomers from the palladium-catalyzed ring-opening reaction of 2 with aryl iodides.
Our group recently investigated the effect of C1 substitution, with ethyl and methoxycarbonyl substituents, on the palladium-catalyzed ring-opening reaction of oxabenzonorbornadiene with aryl iodides (Scheme 4) [15]. We found that the electron-withdrawing methoxycarbonyl substituent on the aryl iodide or bridgehead carbon lowered the yield in all cases and gave aromatized products, while the electron-donating ethyl group on the aryl iodide or bridgehead carbon increased the yield, relative to the unsubstituted parent compound, in all cases. Despite the differences in yield and in the electronic nature of the substituents, only a single regioisomer was obtained through the addition of the aryl group to the olefin carbon furthest from the C1 substituent.
![[1860-5397-12-25-i4]](/bjoc/content/inline/1860-5397-12-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Palladium-catalyzed ring-opening of C1 substituted oxabenzonorbornadiene.
Scheme 4: Palladium-catalyzed ring-opening of C1 substituted oxabenzonorbornadiene.
In this paper, we wanted to apply the arylative ring-opening strategy described above to non-aromatic C1 substituted 7-oxanorbornadiene systems with the goal of understanding its impact on reactivity and regioselectivity. The reaction is expected to follow the same general mechanism as oxabenzonorbornadiene with the aryl group being added to the olefin carbon furthest from the C1 substituent followed by dehydration, producing a novel, highly-substituted, biphenyl derivative.
Results and Discussion
The optimization study focused on the palladium catalyst, Lewis acid additive and solvent, as the requirement for Et3N, zinc and high temperatures had previously been determined in related reactions (Table 1) [15]. C1-methyl-substitued oxanorbornadiene 2a was selected for the optimization study due to the ease at which it can be synthesized in larger quantities.
Table 1: Optimization of palladium catalyst, Lewis acid additive, and solvent.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-25-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Solvent | Lewis Acid | Time (h) | Yield (%)a |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | THF | ZnCl2 | 65 | 9b |
| 2 | Pd(OAc)2, Ph3P | THF | ZnCl2 | 17 | 48b |
| 3 | Pd(PPh3)4 | THF | ZnCl2 | 19 | 58b |
| 4 | PdCl2(PPh3)2 | THF | ZnCl2 | 16 | 88 |
| 5 | PdCl2(PPh3)2 | hexanes | ZnCl2 | 16 | 0 |
| 6 | PdCl2(PPh3)2 | methanol | ZnCl2 | 15 | 25 |
| 7 | PdCl2(PPh3)2 | DCM | ZnCl2 | 16 | 61 |
| 8 | PdCl2(PPh3)2 | toluene | ZnCl2 | 22 | 73 |
| 9 | PdCl2(PPh3)2 | DMF | ZnCl2 | 15 | 85 |
| 10 | PdCl2(PPh3)2 | THF | FeCl3 | 16 | 22 |
| 11 | PdCl2(PPh3)2 | THF | AlCl3 | 16 | 29 |
| 12 | PdCl2(PPh3)2 | THF | ZrCl4 | 16 | 47 |
| 13 | PdCl2(PPh3)2 | THF | ZnI2 | 16 | 48 |
| 14 | PdCl2(PPh3)2 | THF | CuCl2 | 18 | 85 |
aIsolated yield after column chromatography. bYield based on 1H NMR using toluene as internal standard.
The palladium catalysts were selected based on their oxidation state and the presence or absence of phosphine ligands (Table 1, entries 1–4). The catalyst lacking a phosphine ligand gave a low yield and the reaction proceeded very slowly (9%, 65 h, Table 1, entry 1). The addition of phosphine exogenously greatly increased the speed and the yield of the reaction (48%, 17 h, Table 1, entry 2). The palladium catalyst, Pd(PPh3)4, gave a moderate yield (58%, 19 h, Table 1, entry 3), while the palladium(II) catalyst, PdCl2(PPh3)2, gave the best yield of the ring opened product and also reacted the fastest (88%, 16 h, Table 1, entry 4).
A variety of solvents were screened including polar aprotic, polar protic and nonpolar solvents (Table 1, entries 5–9). The nonpolar solvents (hexanes, dichloromethane, and toluene) resulted in a range of yields. There was no conversion to the ring opened product when the reaction was performed in hexanes (Table 1, entry 5), however, DCM and toluene gave much better yields at 61% and 73% respectively (Table 1, entries 7 and 8). The reaction using the polar protic solvent, methanol, gave a low yield (25%, Table 1, entry 6), while the polar aprotic solvents, DMF and THF, gave the best results at 85% and 88% respectively (Table 1, entries 9 and 4).
The Lewis acid additives gave a large range of yields (Table 1, entries 10–14). Iron and aluminium chlorides gave low yields (22%, Table 1, entry 10 and 29%, entry 11), zirconium chloride and zinc iodide gave moderate yields (47%, Table 1, entry 12 and 48%, entry 13) and copper chloride and zinc chloride gave the highest yields (85%, Table 1, entry 14 and 88%, entry 4). The combination of PdCl2(PPh3)2, THF, and zinc chloride resulted in the highest conversion to the ring-opened product.
Once the optimized conditions were found, 2a was reacted with a variety of substituted aryl iodides to illustrate the scope of the reaction with different aryl iodides (Table 2). The effect of the relative position of the substituent to the iodide and the electronic nature of the substituents were investigated. The electron-donating groups, OMe and Me, gave the highest yield in the para position while the electron-withdrawing group, NO2, favoured ring-opening in the meta position. The highest yield for the electron-donating groups was obtained with p-iodoanisole (88%, Table 2, entry 1). Moderate yields were obtained with m- and o-iodoanisoles (62%, Table 2, entry 2 and 69%, entry 3). p-Iodotoluene also gave a high yield (82%, Table 2, entry 4). The yield for m-iodotoluene was close (79%, Table 2, entry 5) but there was a dramatic drop in the yield for o-iodotoluene (30%, Table 2, entry 6), potentially as a result of steric interactions with ortho substituents at site of Pd insertion.
Table 2: Effect of aryl iodide substitution on the ring-opening of oxanorbornadiene.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-25-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | R3 | Time (h) | Product | Yield (%)a |
|---|---|---|---|---|---|---|
| 1 | OMe | H | H | 16 | 3a | 88 |
| 2 | H | OMe | H | 17 | 3b | 62 |
| 3 | H | H | OMe | 17 | 3c | 69 |
| 4 | Me | H | H | 16 | 3d | 82 |
| 5 | H | Me | H | 16 | 3e | 79 |
| 6 | H | H | Me | 19 | 3f | 30 |
| 7 | NO2 | H | H | 16 | 3g | 73 |
| 8 | H | NO2 | H | 16 | 3h | 88 |
| 9 | H | H | NO2 | 19 | 3i | 47 |
aIsolated yield after column chromatography.
The trend seen with electron-donating substituents on the aryl iodide was not seen with the electron-withdrawing substituent, NO2; p-iodonitrobenzene did not give the highest yield for the ring-opened product (73%, Table 2, entry 7). Instead, the highest yield was seen with m-iodonitrobenzene (88%, Table 2, entry 8); the same yield as was seen with p-iodoanisole, and the lowest yield was seen with o-iodonitrobenzene (47%, Table 2, entry 9).
The effect of substitution on the bridgehead carbon of 1 was investigated to determine its impact on the outcome of the ring-opening reaction (Table 3). A wide variety of substituents were chosen, including electron-withdrawing and donating groups, bulky groups, and aromatic rings, in order to examine the impact of steric and electronic changes at the C1 position. The greatest yield for the ring-opened derivative was obtained with the methyl substituent (88%, Table 3, entry 1). When the size of the substituent was increased by one carbon, an ethyl group, the yield decreased to 69% (Table 3, entry 2) and when an even bulkier phenyl group was substituted at the C1 position, the yield was reduced to 33% (Table 3, entry 3). Increasing the steric bulk on the bridgehead carbon appears to impact the reactions’ outcome by hindering access to the site of carbo-palladation. SiMe3 gave a moderate yield of 64% (Table 3, entry 4). The silicon–carbon bond is longer than carbon–carbon bonds, making TMS less bulky than its hydrocarbon counterparts. Both of the electron-withdrawing functional groups, methyl ester, and acetyl, resulted in 0% conversion to the ring-opened product (Table 3, entries 5 and 6). Based on these results, it appears that both electronic and steric interactions at the bridgehead carbon play an important role in the outcome of the reaction.
Table 3: Effect of C1 substitution on the ring-opening reaction of oxanorbornadiene.
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-25-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | OBD | R | Product | Yield (%)a |
|---|---|---|---|---|
| 1 | 2a | Me | 3a | 88 |
| 2 | 2b | Et | 3j | 69 |
| 3 | 2c | Ph | 3k | 33 |
| 4 | 2d | SiMe3 | 3l | 64 |
| 5 | 2e | COOMe | 3m | 0 |
| 6 | 2f | COCH3 | 3n | 0 |
aIsolated yield after column chromatography.
A mechanism for the ring-opening reaction of 2 with aryl iodides has been proposed based on the results obtained (Scheme 5). The reaction begins with the reduction of the palladium(II) catalyst, 4, to palladium(0) 5 by zinc. The Pd(0) catalyst complexes with the aryl iodide 6 forming the palladium-aryl complex 7. This complex undergoes exo selective coordination with 2, which is likely promoted by the Lewis acid ZnCl2 removing the iodide from 7 allowing it to associate with the oxanorbornadiene. This is quickly followed by carbopalladation onto the exposed olefin to give intermediate 8. There are two possible regioisomers, depending on whether the aryl group adds to Ca or Cb. In all cases, only one regioisomer was seen as a result of the addition of the aryl group to Ca. Cleavage of the β-oxygen gives the ring-opened intermediate 9. The palladium species is reduced, producing the final intermediate 10 and regenerating the catalyst. Compound 10 rapidly undergoes base-catalyzed dehydration to give the final product 3.
![[1860-5397-12-25-i5]](/bjoc/content/inline/1860-5397-12-25-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for the palladium-catalyzed ring-opening reaction of oxanorbornadiene.
Scheme 5: Proposed mechanism for the palladium-catalyzed ring-opening reaction of oxanorbornadiene.
Conclusion
In conclusion, we have demonstrated the first examples of palladium-catalyzed ring-opening reactions of 1 with aryl iodides. This reaction was highly regioselective giving only one of the two possible regioisomers via the addition of the aryl group to the less hindered carbon of the olefin (Ca), followed by dehydration, producing novel highly-substituted biphenyl derivatives. We investigated the efficacy of various ring-opening conditions and found that PdCl2(PPh3)2, THF, ZnCl2, Et3N, high temperature, and Zn gave the highest yield.
References
-
Lautens, M. Synlett 1993, 177–185. doi:10.1055/s-1993-22393
Return to citation in text: [1] -
Woo, S.; Keay, B. A. Synthesis 1996, 669–686. doi:10.1055/s-1996-4295
Return to citation in text: [1] -
Chiu, P.; Lautens, M. Using ring-opening reactions of oxabicyclic compounds as a strategy in organic synthesis. In Stereoselective Heterocyclic Synthesis II; Metz, P., Ed.; Topics in Current Chemistry, Vol. 190; Springer: Berlin, Germany, 1997; pp 1–85. doi:10.1007/BFb0119245
Return to citation in text: [1] -
Lautens, M.; Fagnou, K.; Hiebert, S. Acc. Chem. Res. 2003, 36, 48–58. doi:10.1021/ar010112a
Return to citation in text: [1] -
Rayabarapu, D. K.; Cheng, C.-H. Acc. Chem. Res. 2007, 40, 971–983. doi:10.1021/ar600021z
Return to citation in text: [1] -
Li, M.; Yan, X.-X.; Hong, W.; Zhu, X.-Z.; Cao, B.-X.; Sun, J.; Hou, X.-L. Org. Lett. 2004, 6, 2833–2835. doi:10.1021/ol048816i
Return to citation in text: [1] -
Mitscher, L. A. Regiospecific synthesis of anthracyclinone compounds such as daunomycinone. U. S. Patent 4374979 A, Feb 22, 1983.
Return to citation in text: [1] -
Saeidnia, S.; Abdollahi, M. Daru, J. Pharm. Sci. 2013, 21, No. 43. doi:10.1186/2008-2231-21-43
Return to citation in text: [1] -
Lautens, M.; Renaud, J.-L.; Hiebert, S. J. Am. Chem. Soc. 2000, 122, 1804–1805. doi:10.1021/ja993427i
Return to citation in text: [1] -
Imamoto, T.; Sugita, K.; Yoshida, K. J. Am. Chem. Soc. 2005, 127, 11934–11935. doi:10.1021/ja053458f
Return to citation in text: [1] -
Moinet, C.; Fiaud, J.-C. Tetrahedron Lett. 1995, 36, 2051–2052. doi:10.1016/0040-4039(95)00178-F
Return to citation in text: [1] -
Duan, J.-P.; Cheng, C.-H. Tetrahedron Lett. 1993, 34, 4019–4022. doi:10.1016/S0040-4039(00)60605-6
Return to citation in text: [1] -
Duan, J.-P.; Cheng, C.-H. Organometallics 1995, 14, 1608–1618. doi:10.1021/om00004a014
Return to citation in text: [1] -
Lautens, M.; Hiebert, S.; Renaud, J.-L. J. Am. Chem. Soc. 2001, 123, 6834–6839. doi:10.1021/ja010498k
Return to citation in text: [1] -
Raheem, M. A.; Edmunds, M.; Tam, W. Can. J. Chem. 2014, 92, 888–895. doi:10.1139/cjc-2014-0217
Return to citation in text: [1] [2] -
Raheem, M.-A.; Tam, W. Synth. Commun. 2013, 43, 260–267. doi:10.1080/00397911.2011.596640
| 1. | Lautens, M. Synlett 1993, 177–185. doi:10.1055/s-1993-22393 |
| 2. | Woo, S.; Keay, B. A. Synthesis 1996, 669–686. doi:10.1055/s-1996-4295 |
| 3. | Chiu, P.; Lautens, M. Using ring-opening reactions of oxabicyclic compounds as a strategy in organic synthesis. In Stereoselective Heterocyclic Synthesis II; Metz, P., Ed.; Topics in Current Chemistry, Vol. 190; Springer: Berlin, Germany, 1997; pp 1–85. doi:10.1007/BFb0119245 |
| 4. | Lautens, M.; Fagnou, K.; Hiebert, S. Acc. Chem. Res. 2003, 36, 48–58. doi:10.1021/ar010112a |
| 5. | Rayabarapu, D. K.; Cheng, C.-H. Acc. Chem. Res. 2007, 40, 971–983. doi:10.1021/ar600021z |
| 6. | Li, M.; Yan, X.-X.; Hong, W.; Zhu, X.-Z.; Cao, B.-X.; Sun, J.; Hou, X.-L. Org. Lett. 2004, 6, 2833–2835. doi:10.1021/ol048816i |
| 14. | Lautens, M.; Hiebert, S.; Renaud, J.-L. J. Am. Chem. Soc. 2001, 123, 6834–6839. doi:10.1021/ja010498k |
| 9. | Lautens, M.; Renaud, J.-L.; Hiebert, S. J. Am. Chem. Soc. 2000, 122, 1804–1805. doi:10.1021/ja993427i |
| 10. | Imamoto, T.; Sugita, K.; Yoshida, K. J. Am. Chem. Soc. 2005, 127, 11934–11935. doi:10.1021/ja053458f |
| 11. | Moinet, C.; Fiaud, J.-C. Tetrahedron Lett. 1995, 36, 2051–2052. doi:10.1016/0040-4039(95)00178-F |
| 12. | Duan, J.-P.; Cheng, C.-H. Tetrahedron Lett. 1993, 34, 4019–4022. doi:10.1016/S0040-4039(00)60605-6 |
| 13. | Duan, J.-P.; Cheng, C.-H. Organometallics 1995, 14, 1608–1618. doi:10.1021/om00004a014 |
| 8. | Saeidnia, S.; Abdollahi, M. Daru, J. Pharm. Sci. 2013, 21, No. 43. doi:10.1186/2008-2231-21-43 |
| 7. | Mitscher, L. A. Regiospecific synthesis of anthracyclinone compounds such as daunomycinone. U. S. Patent 4374979 A, Feb 22, 1983. |
| 15. | Raheem, M. A.; Edmunds, M.; Tam, W. Can. J. Chem. 2014, 92, 888–895. doi:10.1139/cjc-2014-0217 |
| 15. | Raheem, M. A.; Edmunds, M.; Tam, W. Can. J. Chem. 2014, 92, 888–895. doi:10.1139/cjc-2014-0217 |
© 2016 Edmunds et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)