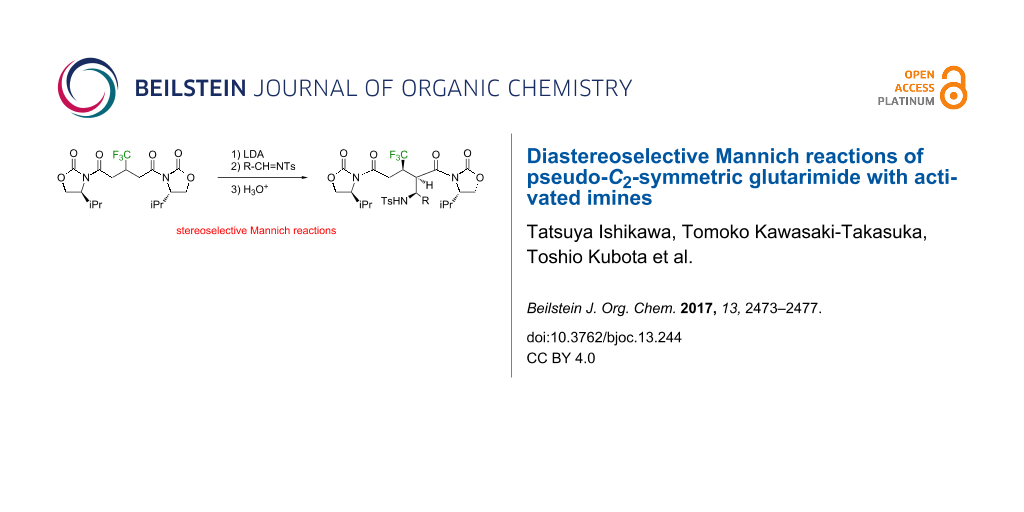
Abstract
As an extension of the boron enolate-based aldol reactions, the oxazolidinone-installed bisimide 1a from 3-(trifluoromethyl)glutaric acid was employed for Mannich reactions with tosylated imines 2 as electrophiles to successfully obtain the corresponding adducts in a stereoselective manner.

Graphical Abstract
Introduction
During these decades, we have been keenly interested in the 3-(trifluoromethyl)glutaric acid derivatives and developed a couple of routes to get successful access to such target molecules with a variety of substituents at the 2-position [1-3]. Previously, the oxazolidinone-installed bisimide 1a was employed for the crossed aldol reactions by the way of boron enolate which allowed the isolation of optically active lactones in good to excellent yields [4,5]. One of the most intriguing features of this protocol is the fact that the enantiomers at the lactone part were readily obtained only by the selection of the tertiary amines employed in the reaction. It is quite apparent that this success is, at least in part, based on its inherent pseudo-C2 symmetric structure [6-8] which enables the formation of plural stereogenic centers by a single operation. These promising results prompted us to extend this aldol protocol to its relative, Mannich reactions [9-13] whose details are reported in this article.
Results and Discussion
On the basis of our previous study [4], the chiral glutarimide 1a was employed as the starting material and optimization of reaction conditions with benzaldehyde-based imines 2 was performed (Table 1). Lithium enolate by the action of LDA to 1a was found to be ineffective as long as the imines with benzyl (2aa) or Boc (2ab) as substituents R were employed (Table 1, entries 1 and 2).
Table 1: Optimization of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-244-i2.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Base | R | Yielda (%) | DSb |
| 1 | THF | LDA | PhCH2 (2aa) | NR | – |
| 2 | THF | LDA | Boc (2ab) | NR | – |
| 3 | THF | LDA | Ts (2ac) | 85 (3a) | 69:18:(13) |
| 4 | THF | NaHMDS | Ts (2ac) | 59 (3a) | 38:43:(19) |
| 5 | THF | LiHMDS | Ts (2ac) | 80 (3a) | 27:62:(11) |
| 6c | DCM | DIPEA | Ts (2ac) | NR | – |
| 7d | THF | LDA | Ts (2ac) | 96 [75] (3a) | 73:17:(10) |
| 8d,e | THF | LDA | Ts (2ac) | 91 (4a) | 73:21:(6) |
aAll yields were determined by 19F NMR and in the bracket isolated yields are given. NR: no reaction. bDiastereoselectivities (DS) were determined by 19F NMR and in the parentheses the sum of the other minor stereoisomers is given. cTiCl4 (3.0 equiv) was used as a Lewis acid. dThe reaction was continued for 6 h. eSubstrate 1b with the phenylglycine-derived oxazolidinone part was employed instead of 1a.
However, the attachment of the stronger electron-withdrawing 4-toluenesulfonyl (Ts) moiety attained efficient activation of the imine 2ac to afford the desired Mannich product 3a in 85% yield as determined by 19F NMR (Table 1, entry 3). NaHMDS (Table 1, entry 4) or LiHMDS (Table 1, entry 5) instead of LDA worked properly affording 3a in a yield of 59 or 80%, respectively. The product 3a theoretically consists of 8 diastereomers and usually 2 isomers predominated with a couple of minor peaks whose sum is given in parentheses (for example, in the case of entry 3 (Table 1), four small peaks were observed in addition to the major two). It is interesting to note that MHMDS (M: Li or Na) and LDA furnished a different isomer as the major product. We have also prepared the titanium enolate [14], but the reaction did not proceed at all (Table 1, entry 6). As the separation of product 3a from unreacted substrate 1a proved to be difficult, the reaction time was extended from 3 h to 6 h. After this time the consumption of 1a was complete and the yield of 3a was almost quantitative (Table 1, entry 7). Because the employment of the phenylalanine-based oxazolidinone as the chiral auxiliary under the same conditions gave similar results as the one obtained from valine (Table 1, entries 8 vs 7), we decided the conditions in entry 7 (Table 1) as the best of all tested and investigated the structural scope of imines 2 at the next stage (Table 2).
Table 2: Scope and limitation of the present Mannich reactions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-244-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Base | R | Time (h) | Yielda (%) | Product | DSb |
| 1 | LDA | Ph (2ac) | 3.0 | 96 [75] | 3a | 73:17:(10) |
| 2 | LiHMDS | Ph (2ac) | 6.0 | 96[74] | 3a | 27:63:(10) |
| 3 | LDA | 4-BrC6H4- (2bc) | 6.0 | 93 [91] | 3b | 62:17:(21) |
| 4 | LiHMDS | 4-BrC6H4- (2bc) | 6.0 | 84 [62] | 3b | 26:61:(13) |
| 5 | LDA | 4-O2NC6H4- (2cc) | 6.0 | quant [92] | 3c | 65:13:(22) |
| 6 | LiHMDS | 4-O2NC6H4- (2cc) | 6.0 | 64 [45] | 3c | 49:44:(7) |
| 7 | LDA | 3-FC6H4- (2dc) | 6.0 | 89 [78] | 3d | 81:6:(13) |
| 8 | LiHMDS | 3-FC6H4- (2dc) | 6.0 | 84 [72] | 3d | 33:51:(16) |
| 9 | LDA | 2-furyl- (2ec) | 8.0 | 95 [55] | 3e | 51:22:(27) |
| 10 | LiHMDS | 2-furyl- (2ec) | 6.0 | 48 | 3e | 28:57:(15) |
| 11 | LDA | 4-MeC6H4- (2fc) | 8.0 | quant [66] | 3f | 51:16:(33) |
| 12 | LiHMDS | 4-MeC6H4- (2fc) | 6.0 | 51 | 3f | 28:64:(8) |
| 13 | LDA | 4-MeOC6H4- (2gc) | 6.0 | 86 | 3g | 34:31:(35) |
| 14 | LiHMDS | 4-MeOC6H4- (2gc) | 6.0 | 26 | 3g | 38:29:(33) |
| 15 | LDA | Et (2hc) | 6.0 | 93 | 3h | 39:36:(25) |
| 16 | LDA | iPr (2ic) | 6.0 | 23 | 3i | 37:17:(46) |
aAll yields were determined by 19F NMR and in the bracket the isolated yields are given. bDiastereoselectivities (DS) were determined by 19F NMR for the crude mixture and in the parentheses the sum of the other minor stereoisomers is given.
As mentioned above, LDA and LiHMDS proved to work in a complementary manner in terms of their diastereoselectivity towards products 3. Therefore, we next tried to use both bases for the reaction with 1a and the resultant enolate was treated with a variety of tosylimines 2 (Table 2). For better overview entry 1 in Table 2 contains the same data as entry 7 in Table 1. It is assumed that the differences between the spectroscopically determined yields to the isolated ones originate from the tedious separation process of the products 3 from the byproduct, 4-toluenesulfonamide (4-MeC6H4SO2NH2) 5 produced by the hydrolysis of the unreacted imines 2 [15]. As expected, aromatic sulfonylimines with electron-withdrawing substituents 2bc–dc acted nicely as electrophiles to furnish the desired adducts 3b–d in high to excellent isolated yields where the complementary formation of the diastereomers was usually observed (Table 2, entries 3–8). Aromatic imines with electron-donating groups, 2fc and 2gc, as well as the heteroaromatic imine 2ec sometimes required longer reaction times, but worked without any significant problems (Table 2, entries 9–14). As a general trend, LiHMDS tended to afford lower chemical yields the reason for which was not clear at present. Imines from aliphatic aldehydes 2hc and 2ic could be used in a similar manner but the steric effect due to the branched structure in the latter affected the reaction to some extent (Table 2, entries 15 and 16). As described above, facile isolation of 3 was sometimes hampered by contamination of the substrate 1a and/or 4-toluenesulfonamide 5 which is noticed by the absence of isolated yields shown in brackets in Table 2.
The configuration of the major diastereomer of 3a (Table 2, entry 1) was unambiguously determined by X-ray crystallographic analysis (Figure 1) [16].
![[1860-5397-13-244-1]](/bjoc/content/figures/1860-5397-13-244-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystallographic analysis of the major diastereomer of 3a (some hydrogen atoms are omitted for clarity). Each color represents the following atoms: gray, carbon; white, hydrogen; blue, nitrogen; red, oxygen; green, fluorine; yellow, sulfur.
Figure 1: Crystallographic analysis of the major diastereomer of 3a (some hydrogen atoms are omitted for clar...
Based on this information a possible reaction mechanism was formulated as depicted in Scheme 1. It is well-known that oxazolidinone-derived imides in general construct bidentate chelation when they are converted to the corresponding metal enolates [17]. The subsequent reactions occur so as to avoid unfavorable steric repulsive interaction with the oxazolidinone substituent, the iPr group in our case (Int-a). The selection of the two enantiotopic enolates should be explained on the basis of the Cieplak effect [18,19] for the enolate conformation with the allylic hydrogen possessing the same plane for making the steric bias minimum (Int-b) [20,21]. The most important argument for this effect is the stabilization of the forming electron-deficient σ*≠ orbital in the transition state by the electron donation from the neighboring orbital. In our case, possible electron donation is expected either by the σC-C in TS(pro-R,si) or σC-CF3 in TS(pro-R,re). Because the former orbital is more electron-rich, imines as electrophiles should approach from the si face of the pro-R enolate (E: an appropriate electrophile in Scheme 1). The same orbital interaction would be operative when the electrophile came closer from the re face of the pro-S enolate which suffered from the existence of the sterically demanding iPr group. As a result, the major reaction pathway was considered to follow the transition state TS(pro-R,si) where the si face of imines seemed to match favorably with a construction of the least sterically demanding conformation, leading to formation of the obtained diastereoisomer 3a.
![[1860-5397-13-244-i1]](/bjoc/content/inline/1860-5397-13-244-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Explanation of the construction of the main stereoisomers.
Scheme 1: Explanation of the construction of the main stereoisomers.
Conclusion
As shown above, the oxazolidinone-installed imide from 3-(trifluoromethyl)glutaric acid 1a was found to afford the corresponding adducts 3 in good to excellent chemical yields in a stereoselective fashion when its enolate was subjected to a solution containing tosylated imines 2 as electrophiles. Further work is going on in this laboratory to utilize the thus obtained adducts 3 and the results will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra for the new compounds, and crystallographic analysis data are available. | ||
| Format: PDF | Size: 539.9 KB | Download |
References
-
Yamazaki, T.; Haga, J.; Kitazume, T.; Nakamura, S. Chem. Lett. 1991, 20, 2171–2174. doi:10.1246/cl.1991.2171
Return to citation in text: [1] -
Yamazaki, T.; Haga, J.; Kitazume, T. Chem. Lett. 1991, 20, 2175–2178. doi:10.1246/cl.1991.2175
Return to citation in text: [1] -
Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013
Return to citation in text: [1] -
Watanabe, Y.; Yamazaki, T.; Kubota, T. Org. Lett. 2010, 12, 268–271. doi:10.1021/ol902592t
Return to citation in text: [1] [2] -
Watanabe, Y.; Yamazaki, T.; Kubota, T. Org. Lett. 2010, 12, 1648. doi:10.1021/ol100413b
Return to citation in text: [1] -
Gao, X.; Han, H.; Krische, M. J. J. Am. Chem. Soc. 2011, 133, 12795–12800. doi:10.1021/ja204570w
Return to citation in text: [1] -
Linclau, B.; Cini, E.; Oake, C. S.; Josse, S.; Light, M.; Ironmonger, V. Angew. Chem., Int. Ed. 2012, 51, 1232–1235. doi:10.1002/anie.201107370
Return to citation in text: [1] -
Ikeuchi, K.; Ido, S.; Yoshimura, S.; Asakawa, T.; Inai, M.; Hamashima, Y.; Kan, T. Org. Lett. 2012, 14, 6016–6019. doi:10.1021/ol302908a
Return to citation in text: [1] -
Arend, M.; Westermann, B.; Risch, N. Angew. Chem., Int. Ed. 1998, 37, 1044–1070. doi:10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E
Return to citation in text: [1] -
Huguenot, F.; Brigaud, T. J. Org. Chem. 2006, 71, 2159–2162. doi:10.1021/jo052323p
Return to citation in text: [1] -
Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635
Return to citation in text: [1] -
Mei, H.; Dai, Y.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Eur. J. Org. Chem. 2014, 2429–2433. doi:10.1002/ejoc.201400118
Return to citation in text: [1] -
Yin, L.; Brewitz, L.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2014, 136, 17958–17961. doi:10.1021/ja511458k
Return to citation in text: [1] -
Franck, X.; Seon-Meniel, B.; Figadère, B. Angew. Chem., Int. Ed. 2006, 45, 5174–5176. doi:10.1002/anie.200600927
Return to citation in text: [1] -
Because of their close Rf values, after evaporation of the volatiles from the chromatographically separated fractions containing these two compounds 3 and 5, this diastereomer mixture was dissolved in an appropriate amount of CH2Cl2 where hexane was added slowly to precipitate the sulfonamide 5.
Return to citation in text: [1] -
CCDC 1575731 contains the supplementary crystallographic data for the major diastereomer of 3a. All data can be acquired free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Evans, D. A.; Ennis, M. D.; Mathre, D. J. J. Am. Chem. Soc. 1982, 104, 1737–1739. doi:10.1021/ja00370a050
Return to citation in text: [1] -
Cieplak, A. S.; Tait, B. D.; Johnson, C. R. J. Am. Chem. Soc. 1989, 111, 8447–8462. doi:10.1021/ja00204a018
Return to citation in text: [1] -
Cieplak, A. S. Chem. Rev. 1999, 99, 1265–1336. doi:10.1021/cr980381n
Return to citation in text: [1] -
Johnson, F. Chem. Rev. 1968, 68, 375–413. doi:10.1021/cr60254a001
Return to citation in text: [1] -
Hoffmann, R. W. Chem. Rev. 1989, 89, 1841–1860. doi:10.1021/cr00098a009
Return to citation in text: [1]
| 1. | Yamazaki, T.; Haga, J.; Kitazume, T.; Nakamura, S. Chem. Lett. 1991, 20, 2171–2174. doi:10.1246/cl.1991.2171 |
| 2. | Yamazaki, T.; Haga, J.; Kitazume, T. Chem. Lett. 1991, 20, 2175–2178. doi:10.1246/cl.1991.2175 |
| 3. | Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013 |
| 4. | Watanabe, Y.; Yamazaki, T.; Kubota, T. Org. Lett. 2010, 12, 268–271. doi:10.1021/ol902592t |
| 9. | Arend, M.; Westermann, B.; Risch, N. Angew. Chem., Int. Ed. 1998, 37, 1044–1070. doi:10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E |
| 10. | Huguenot, F.; Brigaud, T. J. Org. Chem. 2006, 71, 2159–2162. doi:10.1021/jo052323p |
| 11. | Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635 |
| 12. | Mei, H.; Dai, Y.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Eur. J. Org. Chem. 2014, 2429–2433. doi:10.1002/ejoc.201400118 |
| 13. | Yin, L.; Brewitz, L.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2014, 136, 17958–17961. doi:10.1021/ja511458k |
| 6. | Gao, X.; Han, H.; Krische, M. J. J. Am. Chem. Soc. 2011, 133, 12795–12800. doi:10.1021/ja204570w |
| 7. | Linclau, B.; Cini, E.; Oake, C. S.; Josse, S.; Light, M.; Ironmonger, V. Angew. Chem., Int. Ed. 2012, 51, 1232–1235. doi:10.1002/anie.201107370 |
| 8. | Ikeuchi, K.; Ido, S.; Yoshimura, S.; Asakawa, T.; Inai, M.; Hamashima, Y.; Kan, T. Org. Lett. 2012, 14, 6016–6019. doi:10.1021/ol302908a |
| 4. | Watanabe, Y.; Yamazaki, T.; Kubota, T. Org. Lett. 2010, 12, 268–271. doi:10.1021/ol902592t |
| 5. | Watanabe, Y.; Yamazaki, T.; Kubota, T. Org. Lett. 2010, 12, 1648. doi:10.1021/ol100413b |
| 17. | Evans, D. A.; Ennis, M. D.; Mathre, D. J. J. Am. Chem. Soc. 1982, 104, 1737–1739. doi:10.1021/ja00370a050 |
| 20. | Johnson, F. Chem. Rev. 1968, 68, 375–413. doi:10.1021/cr60254a001 |
| 21. | Hoffmann, R. W. Chem. Rev. 1989, 89, 1841–1860. doi:10.1021/cr00098a009 |
| 16. | CCDC 1575731 contains the supplementary crystallographic data for the major diastereomer of 3a. All data can be acquired free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 15. | Because of their close Rf values, after evaporation of the volatiles from the chromatographically separated fractions containing these two compounds 3 and 5, this diastereomer mixture was dissolved in an appropriate amount of CH2Cl2 where hexane was added slowly to precipitate the sulfonamide 5. |
| 14. | Franck, X.; Seon-Meniel, B.; Figadère, B. Angew. Chem., Int. Ed. 2006, 45, 5174–5176. doi:10.1002/anie.200600927 |
| 18. | Cieplak, A. S.; Tait, B. D.; Johnson, C. R. J. Am. Chem. Soc. 1989, 111, 8447–8462. doi:10.1021/ja00204a018 |
| 19. | Cieplak, A. S. Chem. Rev. 1999, 99, 1265–1336. doi:10.1021/cr980381n |
© 2017 Ishikawa et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)