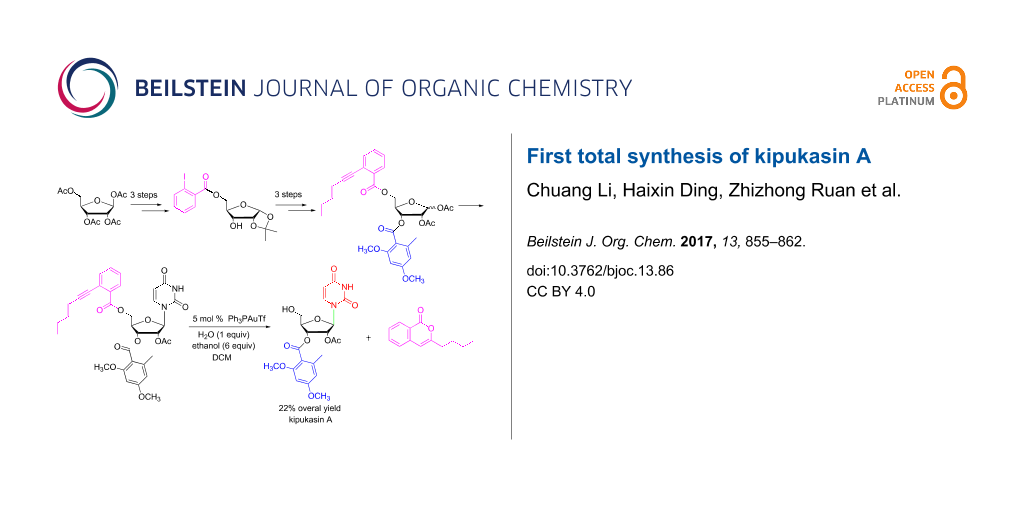
Abstract
In this paper, a practical approach for the total synthesis of kipukasin A is presented with 22% overall yield by using tetra-O-acetyl-β-D-ribose as starting material. An improved iodine-promoted acetonide-forming reaction was developed to access 1,2-O-isopropylidene-α-D-ribofuranose. For the first time, ortho-alkynylbenzoate was used as protecting group for the 5-hydoxy group. After subsequent Vorbrüggen glycosylation, the protecting group could be removed smoothly in the presence of 5 mol % Ph3PAuOTf in dichloromethane to provide kipukasin A in high yield and regioselectivity.

Graphical Abstract
Introduction
Endogenous nucleosides are involved in DNA and RNA synthesis, cell signalling, enzyme regulation and metabolism etc. [1,2]. Therefore, the synthesis of novel nucleosides to mimic their physiological counterparts has potential therapeutic significance, which has led to the development of a large number of antiviral and antitumor drugs [3,4]. On the other hand, naturally occurring nucleosides, especially marine nucleosides, have also played an indispensable role in drug discovery, which make great contribution in the commercialization of cytosine arabinoside (Ara-C), adenine arabinoside (Ara-A) and AZT, etc. [5,6]. Nucleosides and their analogues will continue to play an important role in future drug discovery [7].
In the past decades, exploration of novel naturally occurring marine nucleosides has made expeditious achievements [8-10]. Some of them showed promising antibiotic, antiviral, antiparasitic and antitumor properties. Kipukasins A–G were firstly isolated from solid-substrate fermentation cultures of Hawaiian Aspergillus Versicolor in 2007 (Figure 1) [11]. Later on, kipukasins H, I [12] and J [13] were also isolated from the fungus Aspergillus flavus, which was collected at the South China Sea and the Sea of Okhotsk, respectively.
![[1860-5397-13-86-1]](/bjoc/content/figures/1860-5397-13-86-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Kipukasins are uridine derivatives with unique structural characteristics, which include: (1) a uracil moiety with or without an N-3 methyl group; (2) a 6-methyl-2,4-hydroxy (or methoxy)-benzoyl group at C-2’ or C-3’ position; (3) with or without an acetyl group at 2’-OH position. To the best of our knowledge, they are the first naturally occurring aroyl nucleosides reported up to now. The biological assays showed that kipukasin A owned modest activity against Gram-positive bacteria Staphylococcus aureus (ATCC 29213) [11].
During our ongoing biological studies of marine nucleosides, total syntheses of several marine nucleosides were accomplished in our group [14-18]. In the present paper, we reported a practical approach for the total synthesis of kipukasin A.
Results and Discussion
From the synthetic point of view, it seemed that the most direct approach for the synthesis of kipukasin A was the regioselective modification of commercially available uridine (Figure 2, path a). After carefully assessment, we realized that it would require several steps of protection and deprotection. Especially under alkaline conditions, 2’,3’-transesterification is inevitable to occur in nucleosides [19-21]. The synthetic route would be lengthy and cumbersome. Therefore, a practical total synthesis is in high demand to facilitate the preparation of other kipukasins and their analogues.
![[1860-5397-13-86-2]](/bjoc/content/figures/1860-5397-13-86-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of kipukasin A.
Figure 2: Retrosynthetic analysis of kipukasin A.
The retrosynthetic analysis is shown in Figure 2 (path b). Kipukasin A could be constructed by Vorbrüggen glycosylation [22,23] of a properly protected glycosyl donor 3 with uracil (4). Neighboring group participation of the 2’-O-acetyl group stereoselectively facilitate the β-glycosidic bond formation. Thus, the choice of a suitable protecting group at 5-OH position would be crucial for the success. It should fulfill at least two requirements: (1) it should be stable during the Vorbrüggen glycosylation; and (2) the deprotection process should be performed under very mild and neutral conditions without any influence on the 2’-O-acetyl group. At the same time, ester protection is preferred for Vorbrüggen glycosylations in nucleoside syntheses. Very recently, ortho-alkynylbenzoate was successfully developed by our group as neighboring participation group to synthesize 2’-modified nucleosides [24], which could be removed smoothly in the presence of gold(I) complexes with high yield and selectivity. The conditions are very mild and neutral. In the present paper, we continue to use ortho-alkynylbenzoate as protecting group for the 5’-OH group to fulfill the total synthesis of kipukasin A.
According to the retrosynthetic analysis, we firstly started to synthesis of aroyl building block 9 (Scheme 1). Vilsmeier formylation of 1,3-dihydroxy-5-methylbenzene (5) gave 2,4-dihydroxy-6-methylbenzaldehyde (6) in 75% yield [25,26]. Then compound 6 could react with methyl iodine in acetone by using K2CO3 as base. The obtained 2,4-dimethoxy-6-methylbenzaldehyde (7) was further oxidized with NaH2PO4/NaClO2 in DMSO to provide 2,4-dimethoxy-6-methylbenzoic acid (8) in 81% yield [27,28]. Finally, 2,4-dimethoxy-6-methylbenzoyl chloride (9) was obtained by refluxing with oxalyl chloride in dichloromethane. After removing the solvent and excess oxalyl chloride, 2,4-dimethoxy-6-methylbenzoyl chloride (9) was used directly in the next step without further purification.
![[1860-5397-13-86-i1]](/bjoc/content/inline/1860-5397-13-86-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,4-dimethoxy-6-methylbenzoic chloride. Reagents and conditions: (a) POCl3, DMF, 0 °C to rt, 75%; (b) MeI, K2CO3, acetone, rt, 93%; (c) NaClO2, NaH2PO4, DMSO, rt, 81%; (d) (COCl)2, CH2Cl2, refux.
Scheme 1: Synthesis of 2,4-dimethoxy-6-methylbenzoic chloride. Reagents and conditions: (a) POCl3, DMF, 0 °C ...
Then we started to synthesize glycosylation donor 16 as the key building block (Scheme 2). In previous reports, 3,5-O-diacetyl-1,2-O-isopropylidene-D-ribofuranose (11) was prepared either from D-xylose [29-31] or from tetra-O-acetyl-β-D-ribose (10) [32,33]. In 2009, Koreeda reported an iodine-promoted acetonide-forming reaction of tetra-O-acetyl-β-D-ribose (10) [33]. In this preliminary paper, 25 mol % of iodine was necessary. After systematic optimization, it was found that 6 mol % iodine could complete the reaction efficiently in freshly dried acetone to give 3,5-O-diacetyl-1,2-O-isopropylidene-D-ribofuranose (11) in 88% yield. Then cleavage of the remaining acetyl groups by K2CO3 in MeOH afforded 1,2-O-isopropylidene-D-ribofuranose (12) in 93% yield. Subsequently, the reaction of 1,2-O-isopropylidene-D-ribofuranose (12) with 2-iodobenzoyl chloride (0.9 equiv) gave the corresponding 5-O-benzoyl ester 13 in 80% yield along with a small amount of the 3,5-dibenzoyl ester. The structure of 5-O-benzoyl ester 13 was unambiguously confirmed by X-ray diffraction analysis (Figure 3) [34]. Then Sonogashira cross-coupling with 1-hexyne provided ribose 14 in 78% yield [35].
![[1860-5397-13-86-i2]](/bjoc/content/inline/1860-5397-13-86-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Total synthesis of kipukasin A. Reagents and conditions: (a) I2, acetone, 0 °C to rt, 88%; (b) K2CO3, MeOH, rt, 93%; (c) 2-iodobenzoyl chloride, pyridine, −10 °C to rt, CH2Cl2, 80%; (d) 1-hexyne, PdCl2(PPh3)3, CuI, Et3N, THF, 50 °C , 78%; (e) 9, DMAP, Et3N, CH2Cl2, 0 °C to rt, 74%; (f) Ac2O, H2SO4, acetic acid, rt, 74%; (g) uracil, BSA, TMSOTf, MeCN, 75 °C, 89%; (h) 5% Ph3PAuOTf, H2O, CH2Cl2, EtOH, rt, 90%.
Scheme 2: Total synthesis of kipukasin A. Reagents and conditions: (a) I2, acetone, 0 °C to rt, 88%; (b) K2CO3...
![[1860-5397-13-86-3]](/bjoc/content/figures/1860-5397-13-86-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray structure of compound 13.
Figure 3: X-ray structure of compound 13.
Subsequently, using DMAP as acylation catalyst and triethylamine as base, the former synthesized 2,4-dimethoxy-6-methylbenzoyl chloride (9) reacted with ribose 14 to 3-O-(2,4-dimethoxy-6-methylbenzoyl)ribose 15 in 74% yield. Various spectral analyses (NMR, HPLC) showed no evidence that 2,3-O-transesterification occurred during the esterification reaction. After cleavage of the acetonide group with acetic acid/acetic anhydride/H2SO4, the key glycosylation donor 16 was obtained in 74% yield as a mixture of isomers (α/β = 1:8) [36].
With glycosylation donor 16 in hand, we proceeded to investigate the crucial Vorbrüggen glycosylation with uracil (4). To our delight, in a similar manner as our described in [17], it proved to be efficient to give nucleoside 17 with exclusive β-configuration in 89% yield. At last, using our developed approach [24], kipukasin A was obtained in 90% yield in the presence of 5 mol % Ph3PAuOTf in dichloromethane with H2O (1 equiv) and ethanol (6 equiv). All spectra of the synthetic kipukasin A were consistent with an authentic sample.
Conclusion
In summary, the first total synthesis of kipukasin A was accomplished with 22% overall yield. The reaction sequence includes: (1) an improved iodine-promoted acetonide-forming reaction to synthesize 1,2-O-isopropylidene-D-ribofuranose (12); (2) a Vorbrüggen glycosylation facilitating the preparation for kipukasin derivatives and (3) the first use of ortho-alkynylbenzoate as protecting group of the 5-hydoxy group, which can be removed smoothly in the presence of 5 mol % Ph3PAuOTf in dichloromethane. Biological studies of kipukasin A and the total synthesis of other kipukasin nucleosides by this established approach are ongoing in our group.
Experimental
All reagents and catalysts were purchased from commercial sources (Acros or Aldrich) and used without purification. DCM and CH3CN were dried over CaH2 and distilled prior to use. Et3N was dried over NaH and distilled prior to use. Thin-layer chromatography was performed using silica gel GF-254 plates with detection by UV (254 nm) or charting with 10% sulfuric acid in ethanol. Column chromatography was performed on silica gel (200–300 mesh, Qing-Dao Chemical Company, China). NMR spectra were recorded on a Bruker AV400 spectrometer, and chemical shifts (δ) are reported in ppm. 1H NMR and 13C NMR spectra were calibrated with TMS as internal standard, and coupling constants (J) are reported in Hz. The ESI-HRMS were obtained on a AB SCIEX Triple TOF 4600 spectrometer in positive ion mode. Melting points were measured on an electrothermal apparatus and are uncorrected. Optical rotation values were measured with a Rudolphautopol IV polarimeter.
Synthesis of 3,5-O-diacetyl-1,2-O-isopropylidene-D-ribofuranose (11): To a solution of 1,2,3,5-O-acetyl-β-D-ribofuranose (10, 10.0 g, 31.4 mmol) in dry acetone (100 mL) was added I2 (0.5 g, 2.0 mmol) at 0 °C under argon. After addition, the solution was stirred for 5 h at room temperature and quenched with 40 mL Na2S2O3 (3.0 g, 19.0 mmol). The solvent was evaporated under reduced pressure and distilled water (200 mL) was added to the residue. After that, the solution was extracted with CH2Cl2 (100 mL × 3), the combined organic layer was washed with sat. aq NaHCO3 (100 mL), brine (100 mL), and dried with anhydrous Na2SO4. After filtration, the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (silica gel, PE/EtOAc 1:2, v:v) to afford 11 as a light-yellow oil (7.6 g, 88%). [α]D25 +133.3 (c 0.1, acetone) (lit. [37] [α]D25 +125.9 (c 1.1, CHCl3)); 1H NMR (400 MHz, DMSO-d6) δ 5.81 (d, J = 3.7 Hz, 1H), 4.77 (t, J = 4.2 Hz, 1H), 4.67 (dd, J = 9.1, 4.8 Hz, 1H), 4.24 (dd, J = 12.1, 2.7 Hz, 1H), 4.20–4.15 (m, 1H), 4.05 (dd, J = 12.1, 5.4 Hz, 1H), 2.07 (s, 3H), 2.03 (s, 3H), 1.45 (s, 3H), 1.27 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 170.1, 169.7, 112.2, 103.9, 76.8, 75.1, 71.9, 62.4, 26.4, 20.5, 20.4; LRMS (ESI) m/z: 297.2 [M + Na]+; HRMS (ESI) m/z: [M + Na]+ calcd for C12H18O7Na, 297.0945; found, 297.0943.
Synthesis of 1,2-O-isopropylidene-α-D-ribofuranose (12): The light-yellow oil 11 (7.60 g, 27.7 mmol) was dissolved in MeOH (60 mL). To the solution K2CO3 (0.60 g, 4.4 mmol) was added and the reaction mixture was stirred for 2 h at room temperature. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column to give 12 as a white solid (4.60 g, 93%). Rf 0.30 (CH2Cl2/CH3OH 10:1, v:v); mp 90–91 °C (lit. [38] 87–89 °C); [α]D25 +62.3 (c 0.1, CH3OH) (lit. [38] [α]D25 +49 (c 0.94, CHCl3)); 1H NMR (400 MHz, DMSO-d6) δ 5.65 (d, J = 3.7 Hz, 1H), 4.98 (d, J = 6.7 Hz, 1H), 4.64 (t, J = 5.6 Hz, 1H), 4.43 (t, J = 3.9 Hz, 1H), 3.79–3.56 (m, 3H), 3.41–3.35 (m, 1H), 1.43 (s, 3H), 1.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 111.1, 103.3, 80.3, 79.1, 70.5, 60.2, 26.6, 26.4; LRMS (ESI) m/z: 213.3 [M + Na]+, 189.3 [M − H]−; HRMS (ESI) m/z: [M + Na]+ calcd for C8H13O5Na, 213.0733; found, 213.0731.
Synthesis of 1,2-O-isopropylidene-5-O-(2-iodobenzoyl)-α-D-ribofuranose (13): To a solution of 12 (6.60 g, 34.7 mmol) in dry CH2Cl2 (50 mL) and dry pyridine (7.62 mL) were added 0.2 mL 2-iodobenzoyl chloride (8.4 g, 31.5 mmol) at −10 °C under argon. After addition, the reaction mixture was stirred overnight and quenched with iced water (5 mL). The mixture was washed with sat. NaHCO3 (40 mL), brine (40 mL), and dried over anhydrous MgSO4. After filtration, the filtrated was evaporated to dryness under reduced pressure. The remaining residue was recrystallized by ethanol to obtain 13 as a white powder solid (10.7 g, 80%). Rf 0.35 (PE/EtOAc 2:1, v:v); mp 126–127 °C; [α]D25 +26.86 (c 0.18, CH3OH); 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 1H), 7.82 (dd, J = 7.8, 1.6 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.15 (td, J = 7.7 , 1.6 Hz,1H), 5.84 (d, J = 3.8 Hz, 1H), 4.70 (dd, J = 12.3, 2.5 Hz, 1H), 4.61 (t, J = 4.4 Hz 1H), 4.45 (dd, J = 12.3, 5.2 Hz, 1H), 4.11–4.07 (m, 1H), 3.98 (dd, J = 9.0, 5.1 Hz, 1H), 2.23 (brs, 1H), 1.58 (s, 3H), 1.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.4, 141.4, 134.8, 133.0, 131.4, 128.0, 113.0, 104.2, 94.3, 78.4, 78.3, 72.2, 64.1, 26.7, 26.6; HRMS (ESI) m/z: [M + Na]+ calcd for C15H17IO6Na, 442.9967; found, 442.9980.
Synthesis of 1,2-O-isopropylidene-5-O-(2-(hex-1-yn-1-yl)benzoyl)-α-D-ribofuranose (14): To a solution of 13 (9.0 g, 21.4 mmol) in dry Et3N (25 mL) and dry THF (50 mL) was added CuI (0.41 g, 2.1 mmol), PdCl2(PPh3)3 (2.07 g, 2.1 mmol) and 1-hexyne (2.68 mL, 23.5 mmol). After addition, the reaction mixture was heated at 50 °C for 1 h. TLC detection showed the reaction was finished. The reaction mixture was filtered over a bed of celite. After filtration, the filtrate was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (PE/EtOAc 3:1, v:v) to afford 14 as deep green oil (6.35 g, 78%). Rf 0.43 (PE/EtOAc 2:1,v:v); [α]D25 +21.33 (c 0.15, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 7.8 Hz, 1H), 7.90–7.58 (m, 2H), 7.45 (td, J = 7.2, 2.0 Hz, 1H ), 5.71 (d, J = 3.6 Hz, 1H), 5.34 (d, J = 6.9 Hz, 1H), 4.56–4.50 (m, 2H), 4.22 (dd, J = 12.2, 6.1 Hz, 1H), 4.05–4.01 (m, 1H), 3.86–3.81 (m, 1H), 2.45 (t, J = 6.9 Hz, 2H), 1.55–1.50 (m, 2H), 1.47–1.41 (m, 5H), 1.27 (s, 3H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.6, 133.8, 132.0, 131.9, 129.8, 127.8, 123.3, 111.5, 103.5, 95.9, 78.9, 78.8, 76.9, 71.3, 64.1, 30.1, 26.6, 26.3, 21.4, 18.6, 13.5; HRMS (ESI) m/z: [M + Na]+ calcd for C21H26O6Na, 397.1627; found, 397.1608.
Synthesis of 1,2-O-isopropylidene-3-O-(2,4-dimethoxy-6-methylbenzoyl)-5-O-(2-(hex-1-yn-1-yl)benzoyl)-α-D-ribofuranose (15): To a solution of 14 (3.0 g, 8.0 mmol) in dry CH2Cl2 (25 mL) was added DMAP (97.88 mg, 0.8 mmol) and Et3N (1.05 g, 10.4 mmol). To the mixture benzoyl chloride 9 (2.15 g, 10 mmol) in dry CH2Cl2 (10 mL) was slowly added at 0 °C and stirred overnight at room temperature. The reaction was quenched with methanol (5 mL) and evaporated to dryness under reduced pressure. The obtained residue was dissolved in CH2Cl2 (40 mL), washed with sat. NaHCO3 (40 mL × 2), brine (30 mL × 2), and dried over anhydrous MgSO4. The obtained residue was purified by a silica gel column chromatography (PE/EtOAc 4:1, v:v) to afford 15 as colorless oil (3.3 g, 74%). Rf 0.46 (PE/EtOAc 3:1,v:v); [α]D25 +48.18 (c 0.22, CH3OH); 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.8 Hz, 1H), 7.49 (d, J = 7.2 Hz, 1H), 7.41 (t, J = 7.4 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 6.30 (d, J = 9.7 Hz, 2H), 5.92 (d, J = 3.7 Hz, 1H), 5.03 (t, J = 4.0 Hz, 1H), 4.94 (dd, J = 9.3, 4.8 Hz, 1H), 4.69 (dd, J = 12.2, 2.4 Hz, 1H), 4.56–4.47 (m, 1H), 4.40 (dd, J = 12.2, 5.4 Hz, 1H), 3.79 (s, 3H), 3.76 (s, 3H), 2.47 (t, J = 7.0 Hz, 2H), 2.35 (s, 3H), 1.63–1.57 (m, 2H), 1.55 (s, 3H), 1.47 (d, J = 7.8 Hz, 2H), 1.36 (s, 3H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.3, 166.0, 161.8, 158.8, 139.2, 134.4, 131.7, 131.3, 130.4, 127.1, 124.8, 115.1, 113.1, 106.8, 104.6, 96.4, 96.1, 79.1, 77.3, 75.6, 73.2, 63.4, 55.8, 55.4, 30.7, 26.6, 22.1, 20.1, 19.5, 13.7; HRMS (ESI) m/z: [M + Na]+ calcd for C31H36O9Na, 575.2257; found, 575.2293.
Synthesis of 1,2-O-diacetyl-3-O-(2,4-dimethoxy-6-methylbenzoyl)-5-O-(2-(hex-1-yn-1-yl)benzoyl)-D-ribofuranose (16): A solution of 15 (2.1 g, 3.8 mmol) in acetic acid (10 mL) and Ac2O (1.94 g, 19.0 mmol) was added concentrated sulfuric acid (0.2 mL) dropwise over 10 min. After addition, the reaction mixture was stirred at room temperature for 2 h. TLC detection showed the reaction was finished. The compound was diluted with CH2Cl2 (80 mL) and washed with water (100 mL × 3), sat. NaHCO3 (100 mL × 3), brine (100 mL), and dried (anhydrous Na2SO4). The obtained residue was purified by flash column chromatography to afford 16 as colourless oil (1.68 g, β:α 8:1, 74%). 16-β: Rf 0.30 (PE/EtOAc 4:1, v:v); [α]D25 −10.83 (c 0.23, CH3OH); 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 6.31 (d, J = 2.0 Hz, 1H), 6.29 (d, J = 2.2 Hz, 1H), 6.19 (s, 1H), 5.64 (dd, J = 7.3, 4.9 Hz, 1H), 5.57 (d, J = 4.9 Hz, 1H), 4.72 (dd, J = 12.1, 3.1 Hz, 1H), 4.64–4.57 (m, 1H), 4.41 (dd, J = 12.2, 4.9 Hz, 1H), 3.80 (s, 3H), 3.76 (s, 3H), 2.47 (t, J = 7.1 Hz, 2H), 2.29 (s, 3H), 2.07 (s, 3H), 1.95 (s, 3H), 1.64–1.57 (m, 2H), 1.51–1.44 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.4, 169.2, 167.0, 165.8, 162.0, 158.9, 139.2, 134.5, 131.9, 131.4, 130.4, 127.2, 125.1, 114.7, 107.0, 98.4, 96.7, 96.3, 80.0, 79.1, 74.3, 71.1, 63.9, 55.9, 55.5, 30.8, 22.2, 21.0, 20.7, 20.1, 19.6, 13.8; HRMS (ESI) m/z: [M + Na]+ calcd for C32H36O11Na, 619.2150; found, 619.2147. 16-α: Rf 0.17 (PE/EtOAc 4:1, v:v); [α]D25 +10.20 (c 0.15, CH3OH); 1H NMR (400 MHz, CDCl3) δ 7.89 (dd, J = 7.9, 1.1 Hz, 1H), 7.52 (dd, J = 7.8, 1.0 Hz, 1H), 7.43 (td, J = 7.6, 1.4 Hz, 1H), 7.32 (td, J = 7.7, 1.3 Hz, 1H), 6.48 (d, J = 4.6 Hz, 1H), 6.33– 6.32 (m, 2H), 5.59 (dd, J = 6.8, 3.0 Hz, 1H), 5.40 (dd, J = 6.8, 4.6 Hz, 1H), 4.70–4.66 (m, 1H), 4.65 (dd, J = 12.1, 3.0 Hz, 1H), 4.54 (dd, J = 12.1, 3.7 Hz, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 2.48 (t, J = 7.1 Hz, 2H), 2.36 (s, 3H), 2.06–2.05 (m, 6H), 1.63–1.60 (m, 2H), 1.54–1.43 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.8, 169.5, 167.4, 165.9, 161.9, 158.9, 138.7, 134.5, 131.9, 131.3, 130.2, 127.4, 124.9, 115.3, 106.9, 96.6, 96.4, 94.3, 82.2, 79.1, 70.6, 70.4, 64.2, 56.0, 55.5, 30.8, 22.2, 21.2, 20.5, 20.2, 19.6, 13.8; HRMS (EI) m/z: [M + Na]+ calcd for C32H36O11Na, 619.2150; found, 619.2150.
Synthesis of 1-(2’-O-acetyl-3’-O-(2,4-dimethoxy-6-methylbenzoyl)-5’-O-(2-(hex-1-yn-1-yl)benzoyl)-β-D-ribofuranosyl)uracil (17): To a suspension of uracil (0.24 g, 2.2 mmol) in dry MeCN (15 mL) was added BSA (1.36 g, 6.7 mmol). The mixture was heated at 50 °C for 20 min. After cooled to room temperature, a solution of 16 (1.00 g, 1.7 mmol) in dry MeCN (5 mL) along with TMSOTf (1.30 g, 5.9 mmol) were added to the above reaction mixture at 0 °C. The solution was stirred for 5 min before heating to 75 °C for 3–4 h. Then the reaction mixture was poured into cold sat. NaHCO3 solution (30 mL). It was extracted with CH2Cl2 (50 mL). The combined organic layer was washed with sat. aq NaHCO3 (100 mL × 2), brine (50 mL × 2), and dried with anhydrous Na2SO4. After filtration, the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (DCM/CH3OH, 10:1) to give nucleoside 17 as a white solid (0.96 g, 89%). Rf 0.43 (CH2Cl2/CH3OH 30:1,v:v); mp 69–70 °C; [α]D25 −3.10 (c 0.28, CH3OH); 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1H), 7.88 (d, J = 7.4 Hz, 1H), 7.54 (d, J = 7.4 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.40–7.33 (m, 2H), 6.33 (s, 2H,), 6.19 (d, J = 6.1 Hz, 1H), 5.67–5.64 (m, 1H), 5.52 (d, J = 8.1 Hz, 1H), 5.42 (t, J = 5.9 Hz, 1H), 4.72 (dd, J = 12.4, 2.5 Hz, 1H), 4.64 (dd, J = 12.4, 3.1 Hz, 1H'), 4.58–4.55 (m, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 2.46 (t, J = 7.0 Hz, 2H), 2.32 (s, 3H), 2.05 (s, 3H), 1.63–1.55 (m, 2H), 1.51–1.41 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.7, 166.9, 166.0, 163.1, 162.1, 159.1, 150.5, 139.3, 134.5, 132.2, 131.4, 129.8, 127.6, 124.6, 114.4, 107.1 103.4, 96.8, 96.4, 86.7, 80.6, 78.9, 73.0, 71.1, 63.8, 56.0, 55.5, 30.7, 22.1, 20.6, 20.2, 19.5, 13.7; HRMS (ESI) m/z: [M + Na]+ calcd for C34H36N2O11Na, 671.2217; found, 671.2214.
Synthesis of kipukasin A: To a solution of nucleoside 17 (0.60 g, 0.92 mmol) in dry CH2Cl2 (15 mL) was added H2O (1.0 equiv), and ethanol (6.0 equiv) under an argon atmosphere. The mixture was stirred at room temperature for 20 minutes. A freshly prepared solution of Ph3PAuOTf in CH2Cl2 (5 mol % in 1.0 mL) was added, and stirring was continued at room temperature for 5 hours until nucleoside 17 was consumed as monitored by TLC. The reaction mixture was filtered with celite. After filtration, the filtrate was evaporated to dryness under reduced pressure. The obtained residue was recrystallized in petroleum ether (5 mL) to provide kipukasin A as a white powder solid (387 mg, 90%). Rf 0.36 (CH2Cl2/CH3OH 25:1,v:v); mp 95–96 °C; [α]D25 −37.50 (c 0.12, CH3OH) (lit. [11] [α]D25 −26 (c 0.12, CH3OH)); 1H NMR (400 MHz, CDCl3) δ 9.32 (s, 1H, NH), 7.81 (d, J = 8.2 Hz, 1H, H-6), 6.33 (s, 2H, H-3'', H-5''), 6.14 (d, J = 6.7 Hz, 1H, H-1'), 5.79 (d, J = 8.1 Hz, 1H, H-5), 5.68 (dd, J = 6.4, 2.8 Hz, 1H, H-3'), 5.56 (dd, J = 6.6, 5.7 Hz, 1H, H-2'), 4.33–4.32 (m, 1H, H-4'), 3.96 (s, 2H, H-5'), 3.81 (s, 6H, OMe-2'', OMe-4''), 3.32 (s, 1H, -OH), 2.32 (s, 3H, Me-6''), 2.06 (s, 3H, Me-6'); 13C NMR (101 MHz, CDCl3) δ 170.1 (C-6'), 167.3 (C-7''), 163.4 (C-4), 162.1 (C-2''), 159.1 (C-4''), 150.7 (C-2), 140.9 (C-6), 139.2 (C-6''), 114.7 (C-1''), 107.1 (C-5''), 103.4 (C-5), 96.4 (C-3''), 87.4 (C-1'), 84.0 (C-4'), 73.2 (C-2'), 72.1 (C-3'), 62.2 (C-5'), 56.0 (OMe-4''), 55.5 (OMe-2''), 20.7 (Me-6'), 20.3 (Me-6''); HRMS (ESI) m/z: [M + Na]+ calcd for C21H24N2O10Na, 487.1329; found, 487.1327.
Supporting Information
| Supporting Information File 1: Experimental procedures of compounds 6–9, copies of 1H and 13C NMR spectra of all compounds and X-ray crystal data of compound 13. | ||
| Format: PDF | Size: 1.7 MB | Download |
Acknowledgements
We thank the National Science Foundation of China (no. 21462019 and no. 21676131), the Bureau of Science & Technology of Jiangxi Province (20143ACB20012), the China Postdoctoral Science Foundation (no. 2015M570566) and the Jiangxi Province Postdoctoral Science Foundation (no. 2015KY02) for financial support.
References
-
Roos, W. P.; Kaina, B. Trends Mol. Med. 2006, 12, 440–450. doi:10.1016/j.molmed.2006.07.007
Return to citation in text: [1] -
Hannon, G. J. Nature 2002, 418, 244–251. doi:10.1038/418244a
Return to citation in text: [1] -
De Clercq, E. Med. Res. Rev. 2011, 31, 118–160. doi:10.1002/med.20179
Return to citation in text: [1] -
De Clercq, E. Curr. Opin. Pharmacol. 2010, 10, 507–515. doi:10.1016/j.coph.2010.04.011
Return to citation in text: [1] -
Scheuer, P. J. Med. Res. Rev. 1989, 9, 535–545. doi:10.1002/med.2610090404
Return to citation in text: [1] -
Périgaud, C.; Gosselin, G.; Imbach, J. L. Nucleosides Nucleotides 1992, 11, 903–945. doi:10.1080/07328319208021748
Return to citation in text: [1] -
Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010
Return to citation in text: [1] -
Huang, R.-M.; Chen, Y.-N.; Zeng, Z.; Gao, C.-H.; Su, X.; Peng, Y. Mar. Drugs 2014, 12, 5817–5838. doi:10.3390/md12125817
Return to citation in text: [1] -
Blunt, J. W.; Copp, B. R.; Keyzers, R. A.; Munro, M. H. G.; Prinsep, M. R. Nat. Prod. Rep. 2016, 33, 382–431. doi:10.1039/C5NP00156K
Return to citation in text: [1] -
Bhakuni, D. S.; Rawat, D. R. Bioactive Marine Nucleosides. Bioactive Marine Natural Products; Springer Netherlands: Dordrecht, 2006; pp 208–234. doi:10.1007/1-4020-3484-9_8
Return to citation in text: [1] -
Jiao, P.; Mudur, S. V.; Gloer, J. B.; Wicklow, D. T. J. Nat. Prod. 2007, 70, 1308–1311. doi:10.1021/np070241l
Return to citation in text: [1] [2] [3] -
Chen, M.; Fu, X.-M.; Kong, C.-J.; Wang, C.-Y. Nat. Prod. Res. 2014, 28, 895–900. doi:10.1080/14786419.2014.891114
Return to citation in text: [1] -
Zhuravleva, O. I.; Kirichuk, N. N.; Denisenko, V. A.; Dimtrenok, P. S.; Pivkin, M. V.; Afiyatullov, S. S. Chem. Nat. Compd. 2016, 2, 266–268. doi:10.1007/s10600-016-1610-y
Return to citation in text: [1] -
Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. Mar. Drugs 2012, 10, 881–889. doi:10.3390/md10040881
Return to citation in text: [1] -
Song, Y.; Yang, R.; Ding, H.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1213–1218. doi:10.1055/s-0030-1259961
Return to citation in text: [1] -
Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. Chin. Chem. Lett. 2013, 24, 379–382. doi:10.1016/j.cclet.2013.03.014
Return to citation in text: [1] -
Ding, H.; Li, W.; Ruan, Z.; Yang, R.; Miao, Z.; Xiao, Q.; Wu, J. Beilstein J. Org. Chem. 2014, 10, 1681–1685. doi:10.3762/bjoc.10.176
Return to citation in text: [1] [2] -
Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1442–1446. doi:10.1055/s-0030-1259975
Return to citation in text: [1] -
Jackson, M. D.; Denu, J. M. J. Biol. Chem. 2002, 277, 18535–18544. doi:10.1074/jbc.M200671200
Return to citation in text: [1] -
Hasegawa, H.; Akira, K.; Shinohara, Y.; Kasuya, Y.; Hashimoto, T. Biol. Pharm. Bull. 2001, 24, 852–855. doi:10.1248/bpb.24.852
Return to citation in text: [1] -
Dvorakova, M.; Pribylova, M.; Pohl, R.; Migaud, M. E.; Vanek, T. Tetrahedron 2012, 68, 6701–6711. doi:10.1016/j.tet.2012.05.117
Return to citation in text: [1] -
Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley Sons Inc.: New York, USA, 2001.
Return to citation in text: [1] -
Niedballa, U.; Vorbrüggen, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 461–462. doi:10.1002/anie.197004612
Return to citation in text: [1] -
Ding, H.; Li, C.; Zhou, Y.; Hong, S.; Zhang, N.; Xiao, Q. RSC Adv. 2017, 7, 1814–1817. doi:10.1039/C6RA27790J
Return to citation in text: [1] [2] -
Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Org. Lett. 2003, 5, 4481–4484. doi:10.1021/ol030109m
Return to citation in text: [1] -
Xie, L.; Takeuchi, Y.; Cosentino, L. M.; McPhail, A. T.; Lee, K.-H. J. Med. Chem. 2001, 44, 664–671. doi:10.1021/jm000070g
Return to citation in text: [1] -
Solladié, G.; Rubio, A.; Carreño, M. C.; Ruano, J. L. G. Tetrahedron: Asymmetry 1990, 1, 187–198. doi:10.1016/0957-4166(90)90013-Z
Return to citation in text: [1] -
Wang, P.; Zhang, Z.; Yu, B. J. Org. Chem. 2005, 70, 8884–8889. doi:10.1021/jo051384k
Return to citation in text: [1] -
Koth, D.; Fiedler, A.; Scholz, S.; Gottschaldt, M. J. Carbohydr. Chem. 2007, 26, 267–278. doi:10.1080/07328300701540175
Return to citation in text: [1] -
Kim, J.; Weledji, Y. N.; Greenberg, M. M. J. Org. Chem. 2004, 69, 6100–6104. doi:10.1021/jo049033d
Return to citation in text: [1] -
Gosselin, G.; Puech, F.; Génu-Dellac, C.; Imbach, J.-L. Carbohydr. Res. 1993, 249, 1–17. doi:10.1016/0008-6215(93)84056-C
Return to citation in text: [1] -
More, J. D.; Campbell, M. G. Tetrahedron Lett. 2009, 50, 2617–2619. doi:10.1016/j.tetlet.2009.03.116
Return to citation in text: [1] -
Houston, T. A.; Koreeda, M. Carbohydr. Res. 2009, 344, 2240–2244. doi:10.1016/j.carres.2009.08.026
Return to citation in text: [1] [2] -
Crystallographic data for compound 13, C15H17IO6, M = 420.18, crystal dimensions 0.26 × 0.28 × 0.30 mm, orthorhombic, space group, P212121 (No. 19). CCDC 1524599 contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Date Centre via http://www.ccdc.cam.ac.uk/ date_request/cif.
Return to citation in text: [1] -
Sonogashira, K. Palladium-Catalyzed Alkynylation: Sonogashira Alkyne Synthesis. In Handbook of organopalladium chemistry for organic synthesis; Negishi, E.-i., Ed.; John Wiley & Sons Inc.: New York, 2002; pp 493–529. doi:10.1002/0471212466.ch22
Return to citation in text: [1] -
Mathé, C.; Imbach, J.-L.; Gosselin, G. Carbohydr. Res. 1999, 323, 226–229. doi:10.1016/S0008-6215(99)00267-0
Return to citation in text: [1] -
Jiangseubchatveera, N.; Bouillon, M. E.; Liawruangrath, B.; Liawruangrath, S.; Nash, R. J.; Pyne, S. G. Org. Biomol. Chem. 2013, 11, 3826–3833. doi:10.1039/c3ob40374b
Return to citation in text: [1] -
Lenagh-Snow, G. M. J.; Araujo, N.; Jenkinson, S. F.; Rutherford, C.; Nakagawa, S.; Kato, A.; Yu, C.-Y.; Weymouth-Wilson, A. C.; Fleet, G. W. J. Org. Lett. 2011, 13, 5834–5837. doi:10.1021/ol2024482
Return to citation in text: [1] [2]
| 1. | Roos, W. P.; Kaina, B. Trends Mol. Med. 2006, 12, 440–450. doi:10.1016/j.molmed.2006.07.007 |
| 2. | Hannon, G. J. Nature 2002, 418, 244–251. doi:10.1038/418244a |
| 8. | Huang, R.-M.; Chen, Y.-N.; Zeng, Z.; Gao, C.-H.; Su, X.; Peng, Y. Mar. Drugs 2014, 12, 5817–5838. doi:10.3390/md12125817 |
| 9. | Blunt, J. W.; Copp, B. R.; Keyzers, R. A.; Munro, M. H. G.; Prinsep, M. R. Nat. Prod. Rep. 2016, 33, 382–431. doi:10.1039/C5NP00156K |
| 10. | Bhakuni, D. S.; Rawat, D. R. Bioactive Marine Nucleosides. Bioactive Marine Natural Products; Springer Netherlands: Dordrecht, 2006; pp 208–234. doi:10.1007/1-4020-3484-9_8 |
| 27. | Solladié, G.; Rubio, A.; Carreño, M. C.; Ruano, J. L. G. Tetrahedron: Asymmetry 1990, 1, 187–198. doi:10.1016/0957-4166(90)90013-Z |
| 28. | Wang, P.; Zhang, Z.; Yu, B. J. Org. Chem. 2005, 70, 8884–8889. doi:10.1021/jo051384k |
| 7. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 29. | Koth, D.; Fiedler, A.; Scholz, S.; Gottschaldt, M. J. Carbohydr. Chem. 2007, 26, 267–278. doi:10.1080/07328300701540175 |
| 30. | Kim, J.; Weledji, Y. N.; Greenberg, M. M. J. Org. Chem. 2004, 69, 6100–6104. doi:10.1021/jo049033d |
| 31. | Gosselin, G.; Puech, F.; Génu-Dellac, C.; Imbach, J.-L. Carbohydr. Res. 1993, 249, 1–17. doi:10.1016/0008-6215(93)84056-C |
| 5. | Scheuer, P. J. Med. Res. Rev. 1989, 9, 535–545. doi:10.1002/med.2610090404 |
| 6. | Périgaud, C.; Gosselin, G.; Imbach, J. L. Nucleosides Nucleotides 1992, 11, 903–945. doi:10.1080/07328319208021748 |
| 24. | Ding, H.; Li, C.; Zhou, Y.; Hong, S.; Zhang, N.; Xiao, Q. RSC Adv. 2017, 7, 1814–1817. doi:10.1039/C6RA27790J |
| 3. | De Clercq, E. Med. Res. Rev. 2011, 31, 118–160. doi:10.1002/med.20179 |
| 4. | De Clercq, E. Curr. Opin. Pharmacol. 2010, 10, 507–515. doi:10.1016/j.coph.2010.04.011 |
| 25. | Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Org. Lett. 2003, 5, 4481–4484. doi:10.1021/ol030109m |
| 26. | Xie, L.; Takeuchi, Y.; Cosentino, L. M.; McPhail, A. T.; Lee, K.-H. J. Med. Chem. 2001, 44, 664–671. doi:10.1021/jm000070g |
| 11. | Jiao, P.; Mudur, S. V.; Gloer, J. B.; Wicklow, D. T. J. Nat. Prod. 2007, 70, 1308–1311. doi:10.1021/np070241l |
| 19. | Jackson, M. D.; Denu, J. M. J. Biol. Chem. 2002, 277, 18535–18544. doi:10.1074/jbc.M200671200 |
| 20. | Hasegawa, H.; Akira, K.; Shinohara, Y.; Kasuya, Y.; Hashimoto, T. Biol. Pharm. Bull. 2001, 24, 852–855. doi:10.1248/bpb.24.852 |
| 21. | Dvorakova, M.; Pribylova, M.; Pohl, R.; Migaud, M. E.; Vanek, T. Tetrahedron 2012, 68, 6701–6711. doi:10.1016/j.tet.2012.05.117 |
| 13. | Zhuravleva, O. I.; Kirichuk, N. N.; Denisenko, V. A.; Dimtrenok, P. S.; Pivkin, M. V.; Afiyatullov, S. S. Chem. Nat. Compd. 2016, 2, 266–268. doi:10.1007/s10600-016-1610-y |
| 22. | Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley Sons Inc.: New York, USA, 2001. |
| 23. | Niedballa, U.; Vorbrüggen, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 461–462. doi:10.1002/anie.197004612 |
| 12. | Chen, M.; Fu, X.-M.; Kong, C.-J.; Wang, C.-Y. Nat. Prod. Res. 2014, 28, 895–900. doi:10.1080/14786419.2014.891114 |
| 11. | Jiao, P.; Mudur, S. V.; Gloer, J. B.; Wicklow, D. T. J. Nat. Prod. 2007, 70, 1308–1311. doi:10.1021/np070241l |
| 14. | Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. Mar. Drugs 2012, 10, 881–889. doi:10.3390/md10040881 |
| 15. | Song, Y.; Yang, R.; Ding, H.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1213–1218. doi:10.1055/s-0030-1259961 |
| 16. | Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. Chin. Chem. Lett. 2013, 24, 379–382. doi:10.1016/j.cclet.2013.03.014 |
| 17. | Ding, H.; Li, W.; Ruan, Z.; Yang, R.; Miao, Z.; Xiao, Q.; Wu, J. Beilstein J. Org. Chem. 2014, 10, 1681–1685. doi:10.3762/bjoc.10.176 |
| 18. | Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1442–1446. doi:10.1055/s-0030-1259975 |
| 34. | Crystallographic data for compound 13, C15H17IO6, M = 420.18, crystal dimensions 0.26 × 0.28 × 0.30 mm, orthorhombic, space group, P212121 (No. 19). CCDC 1524599 contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Date Centre via http://www.ccdc.cam.ac.uk/ date_request/cif. |
| 32. | More, J. D.; Campbell, M. G. Tetrahedron Lett. 2009, 50, 2617–2619. doi:10.1016/j.tetlet.2009.03.116 |
| 33. | Houston, T. A.; Koreeda, M. Carbohydr. Res. 2009, 344, 2240–2244. doi:10.1016/j.carres.2009.08.026 |
| 33. | Houston, T. A.; Koreeda, M. Carbohydr. Res. 2009, 344, 2240–2244. doi:10.1016/j.carres.2009.08.026 |
| 38. | Lenagh-Snow, G. M. J.; Araujo, N.; Jenkinson, S. F.; Rutherford, C.; Nakagawa, S.; Kato, A.; Yu, C.-Y.; Weymouth-Wilson, A. C.; Fleet, G. W. J. Org. Lett. 2011, 13, 5834–5837. doi:10.1021/ol2024482 |
| 11. | Jiao, P.; Mudur, S. V.; Gloer, J. B.; Wicklow, D. T. J. Nat. Prod. 2007, 70, 1308–1311. doi:10.1021/np070241l |
| 37. | Jiangseubchatveera, N.; Bouillon, M. E.; Liawruangrath, B.; Liawruangrath, S.; Nash, R. J.; Pyne, S. G. Org. Biomol. Chem. 2013, 11, 3826–3833. doi:10.1039/c3ob40374b |
| 38. | Lenagh-Snow, G. M. J.; Araujo, N.; Jenkinson, S. F.; Rutherford, C.; Nakagawa, S.; Kato, A.; Yu, C.-Y.; Weymouth-Wilson, A. C.; Fleet, G. W. J. Org. Lett. 2011, 13, 5834–5837. doi:10.1021/ol2024482 |
| 17. | Ding, H.; Li, W.; Ruan, Z.; Yang, R.; Miao, Z.; Xiao, Q.; Wu, J. Beilstein J. Org. Chem. 2014, 10, 1681–1685. doi:10.3762/bjoc.10.176 |
| 24. | Ding, H.; Li, C.; Zhou, Y.; Hong, S.; Zhang, N.; Xiao, Q. RSC Adv. 2017, 7, 1814–1817. doi:10.1039/C6RA27790J |
| 35. | Sonogashira, K. Palladium-Catalyzed Alkynylation: Sonogashira Alkyne Synthesis. In Handbook of organopalladium chemistry for organic synthesis; Negishi, E.-i., Ed.; John Wiley & Sons Inc.: New York, 2002; pp 493–529. doi:10.1002/0471212466.ch22 |
| 36. | Mathé, C.; Imbach, J.-L.; Gosselin, G. Carbohydr. Res. 1999, 323, 226–229. doi:10.1016/S0008-6215(99)00267-0 |
© 2017 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)