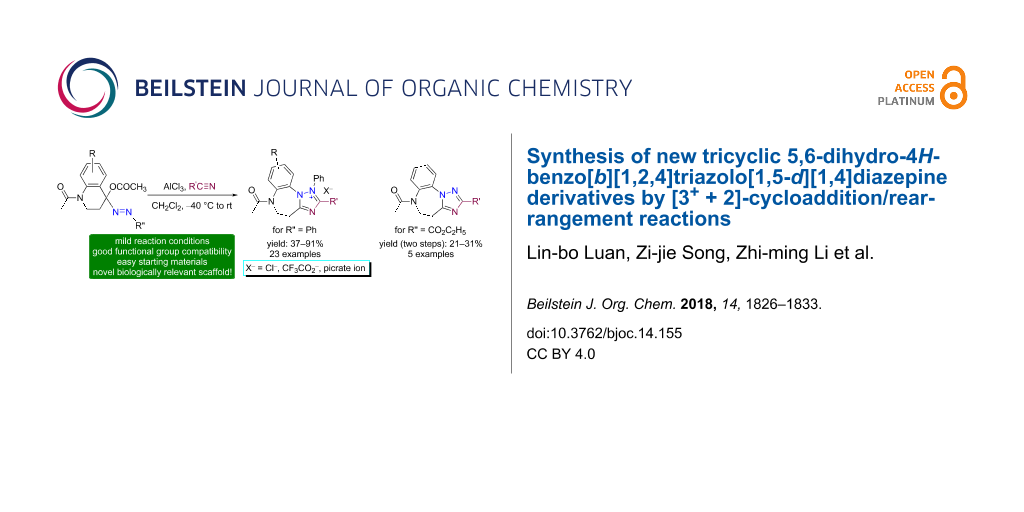
Abstract
Two new series of tricyclic heterocycles, namely 5,6-dihydro-4H-benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10 and the related neutral, free bases 13 were synthesized from 4-acetoxy-1-acetyl-4-phenylazo-1,2,3,4-tetrahydroquinolines 8 and nitriles 9 in the presence of aluminium chloride by the [3+ + 2]-cycloaddition reaction of the in situ generated azocarbenium intermediates 14 followed by a ring-expansion rearrangement. In the rearrangement reaction, the phenyl substituent in the initially formed spiro-triazolium adducts 16 underwent a [1,2]-migration from C(3) to the electron-deficient N(2). This led to the ring expansion from 6-membered piperidine to 7-membered diazepine furnishing the tricyclic 1,2,4-triazole-fused 1,4-benzodiazepines.

Graphical Abstract
Introduction
Heterocyclic compounds comprising a 1,4-benzodiazepine (BDZ) ring have been a topic of continued interest as they exhibit a wide spectrum of drug-like profiles such as good anticonvulsant activity against acutely elicited seizure, particularly for the central nervous system [1-5]. Many biologically active small molecules with such a core structure have been marketed for the treatment of various diseases, mostly as psychotropic substances [6]. Thus, for example, chlordiazepoxide (Librium) and the benzodiazepine diazepam (Valium) have been a sedative and hypnotic medication and marked for the treatment of anxiety, insomnia and withdrawal symptoms from alcohol and/or drug abuse by Hoffmann-La Roche since the 1960’s. Benzodiazepines are thus categorized as a privileged heterocyclic system that is the structural basis of a large number of drugs as defined by Evans about 30 years ago [7-12]. Later, more detailed research revealed that improved biological activities, metabolic stability and other profiles could be achieved when a third heterocycle, especially a 1,2,4-triazolo moiety, was attached to the seven-membered ring as part of 1,4-benzodiazepine [13,14]. Among various reported 1,2,4-triazole-annulated 1,4-benzodiazepines, triazolam (I), estazolam (II), alprazolam (III) and pyrazolam (IV) are prominent examples of such clinically drugs having enhanced effects on the neurotransmitter γ-aminobutyric acid (GABA) at the GABAA receptor and low toxicity (Figure 1) [15].
![[1860-5397-14-155-1]](/bjoc/content/figures/1860-5397-14-155-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of marketed pharmaceutical 1,2,4-triazolobenzodiazepines.
Figure 1: Examples of marketed pharmaceutical 1,2,4-triazolobenzodiazepines.
Although 1,4-benzodiazepines are widely prescribed medicines, side effects like drowsiness, drug resistance, addiction and withdrawal potential are detrimental [16]. Consequently, the development of expedient synthetic protocols to access new members of 1,4-benzodiazepine derivatives has long been the subject of considerable interest aiming at the discovery of biologically active and drug-like compounds [17-30].
Over the past years, we have been engaged in the synthesis of novel 1,2,4-triazolo heterocycles annulated to benzoazepine or heteroazepine derivatives by a tandem [3+ + 2]-cycloaddition/rearrangement between 1-aza-2-azoniaallenium ions with nitriles [31-34]. In the [3+ + 2]-cycloaddition, α-acetoxyazo intermediates are initially transformed into an azocarbenium ion. Subsequently, it takes part in a cationic cycloaddition reaction with the triple bond of a nitrile followed by a rearrangement reaction [35-39]. This type of ionic cycloaddition–rearrangement protocol proved to be quite general and has also been conducted intramolecularly for a tethered olefin moiety by Brewer and co-workers in recent years [40-42]. In the present work, motivated to achieve structural diversity and potential biological profile improvement of 1,4-benzodiazepines, we wish to describe the synthesis of unprecedented tricyclic heterocycles, i.e., 5,6-dihydro-4H-benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10 and the related neutral free base derivatives 13 via the cationic [3+ + 2]-cycloaddition/rearrangement reactions using the bicyclic 4-acetoxy-4-azo-1,2,3,4-tetrahydroquinolines 8/12 as the key starting material.
Results and Discussion
The required N-acylated 2,3-dihydro-4(1H)-quinolones 6 were generally prepared following a literature method with the acid-catalysed Fries rearrangement of N-arylazetidin-2-ones of the general form 4 [43,44]. As illustrated in Scheme 1, the preparation starts from the related anilines 1 which were acylated with 3-bromopropionyl chloride (2) to afford amides 3. Upon basic treatment with t-BuONa, the amides 3 were converted to the cyclized β-propiolactams 4. In the presence of triflic acid, the Fries rearrangement occurred smoothly to yield the dihydroquinolinones 5, which were then converted to 1-acetyl-2,3-dihydroquinolin-4(1H)-ones 6 by reaction with acetyl chloride in 36–56% overall yields (Scheme 1, see Supporting Information File 1 for details).
![[1860-5397-14-155-i1]](/bjoc/content/inline/1860-5397-14-155-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of N-acylated 2,3-dihydro-4(1H)-quinolones 6.
Scheme 1: Preparation of N-acylated 2,3-dihydro-4(1H)-quinolones 6.
The phenylhydrazones 7 were readily acquired by condensation of the quinolones 6 and phenylhydrazine with a catalytic amount of AcOH in refluxing n-propyl alcohol. Subsequently, the hydrazones 7 were converted into the 4-acetoxy-1-acetyl-4-phenylazo-1,2,3,4-tetrahydroquinolines 8 via the oxidation with hypervalent iodine(III) reagent PhI(OAc)2 (Scheme 2) [45]. The electron-withdrawing N-acetyl functionality in compounds 8 was introduced to conceal the basicity of the N(1) atom, thus avoiding the plausible disturbance in the following cationic polar cycloaddition/rearrangement reaction.
![[1860-5397-14-155-i2]](/bjoc/content/inline/1860-5397-14-155-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of α-acetoxyazo compounds 8a–g. Reaction conditions: for synthesis of 8a: 7a (10.42 mmol), PhI(OAc)2 (12.50 mmol), AcOH (10 mL); for synthesis of 8b–g: 7b–g (0.71 mmol), PhI(OAc)2 (0.85 mmol), AcOH (1 mL). bIsolated yield. cRatio of E/Z isomers was not determined.
Scheme 2: Synthesis of α-acetoxyazo compounds 8a–g. Reaction conditions: for synthesis of 8a: 7a (10.42 mmol)...
After the accomplishment of the synthesis of key intermediates 8, we tried to apply the well-documented protocol in this laboratory for the synthesis of the target tricyclic heterocycles [46]. Thus, the α-acetoxyazo compound 8a was allowed to react with acetonitrile in the presence of AlCl3 as a Lewis acid at low temperature (−40 °C) in dry CH2Cl2 for a period of 0.5 h. Then the reaction mixture was gradually warmed to room temperature and kept at this temperature for an additional hour. The work-up afforded successfully the 5,6-dihydro-4H-benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salt 10a as a white solid in 81% yield. It is noteworthy to mention that the counter ion in the product had been changed from anion AlCl3(OAc)− to picrate anion. This variation proved to be quite beneficial for the formation of stable salts.
Encouraged by the above success, we then examined the protocol’s efficacy by applying different kinds of nitriles. As can be seen from Scheme 3, most of the expected tricyclic benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10 were produced readily from the corresponding acetoxyazo compounds 8 and the corresponding nitriles 9. Moderate to good yields of 10a–g were obtained with aliphatic nitriles, except for 2-chloroacetonitrile which gave a low yield (37%) of 10f from the reaction with 8a. This type of reaction worked also well with 3-methoxypropanenitrile to provide salt 10e in 88% yield. Similarly, 2-phenylacetonitrile participated in the reaction smoothly to give 10g in 73% yield.
![[1860-5397-14-155-i3]](/bjoc/content/inline/1860-5397-14-155-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of tricyclic benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10. Reaction conditions: substrate 8 (0.25 mmol) in CH2Cl2 (2 mL), nitrile 9 (0.35 mmol), AlCl3 (0.35 mmol), CH2Cl2 (5 mL), −40 °C for 0.5 h, then room temperature for 1 h under an atmosphere of N2. bIsolated yields.
Scheme 3: Synthesis of tricyclic benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10. Reaction conditions...
As mentioned above, with the purpose to obtain stable tricyclic 1,2,4-triazolodiazepinium salts 10, the anion AlCl3(OAc)− in the initially formed products was replaced with a suitable counter ion such as chloride, trifluoroacetate and picrate, which proved to be a useful way to facilitate the isolation of the target salts 10.
In order to further explore the scope and generality in view of nitriles, the strategy was also probed with aromatic nitriles 9 that were reacted with 8 under the same reaction conditions. The nucleophilicity of the N atom in aromatic nitriles should be lower than that of aliphatic ones owing to the conjugation of the triple bond with the benzene ring. To our delight, compound 8a reacted smoothly with benzonitrile to give the 2-phenyl-substituted benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium picrate 10h in 83% yield. Then the effect of the electronic nature of benzonitriles was examined with benzonitriles carrying different functionalities. It was disclosed that various electron-donating and electron-withdrawing groups on the benzene ring were compatible with the reaction conditions furnishing the desired products 10i–r in good yields. This indicated that the nucleophilicity of the nitrogen atom of nitrile 9 is strong enough to override the electronic effect of the substituent in the benzene ring. However, when nicotinonitrile was employed, the reaction was unsuccessful, and the expected product 10s could not be identified in the reaction mixture. We speculate that this could be due to the stronger electron density at the N atom in the pyridine ring that interferes with the action of the Lewis acid.
By employment of 8a as a model substrate, we next turned our attention to determine possible steric effects with o-, m- and p-methyl-substituted benzonitriles. The tested reactions proceeded well to give the products 10t–v with comparable good yields, respectively. This indicated that there was no obvious steric impact on the reaction with monosubstituted benzonitriles. On the other hand, from disubstituted benzonitriles including 2-fluoro-4-methylbenzonitrile and 2-methyl-4-chlorobenzonitrile, the tricyclic benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10w and 10x were obtained in moderate yields of 76% and 67%, respectively. However, one limitation was observed for the reaction between 8a and 2,6-dimethylbenzonitrile in which the cyano group is flanked by two substituents in the ortho-positions. Under the same conditions, the reaction failed to give the desired product 10y. This result could be attributed to the increased steric hindrance in nitrile moiety.
As can be seen from Scheme 3, equally satisfactory results could be obtained from the reaction of other acetoxyazo compounds 8 with several different substituents on the benzene ring, as exemplified by the synthesis of 10b,c,l–o. These results demonstrated the broad scope of the present strategy for the preparation of novel tricyclic heterocycles 10. Based on the success in the synthesis of the triazolobenzodiazepinium salts 10, we then turned our efforts to access the N(1)-unsubstituted neutral tricyclic heterocycles. To this end, the phenylhydrazones 7 were replaced by the ethoxycarbonylhydrazone 11, in which the ethoxycarbonyl group was hoped to be removable by hydrolysis [34]. As presented in Scheme 4, ethyl carbazate was used to prepare the hydrazone 11 by condensation with 2,3-dihydro-4(1H)-quinolone 6a [46]. However, it was odd that the oxidation using the hypervalent iodine(III) reagent PhI(OAc)2 as described for phenylhydrazones 7 failed to produce the expected α-acetoxy-ethoxycarbonyl compound 12. Instead, the hydrazone 11 remained intact and was recovered. Therefore, we switched to a stronger oxidant, Pb(OAc)4. To our pleasure, hydrazone 11 was successfully oxidized to provide the required azoester 12. However, NMR analysis revealed that compound 12 was impure and contained also some cyclized byproduct [47]. Furthermore, it was discovered that compound 12 was quite unstable and tended to degrade when it was subjected to chromatographic separation. Based on this, we planned to use the product mixture resulting from the oxidation of hydrazone 11 directly for the following reaction with nitriles 9 in the presence of AlCl3. We were pleased to observe that the reaction proceeded as expected under the usual reaction conditions and the desired products 13 were obtained after quenching with H2O at 0 °C. As can be seen from Scheme 4, both aliphatic and aromatic nitriles worked similarly.
![[1860-5397-14-155-i4]](/bjoc/content/inline/1860-5397-14-155-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N(1)-unsubstituted benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepines 13. Reaction conditions: a) compound 11 (1.20 mmol), Pb(OAc)4 (1.44 mmol), CH2Cl2 (10 mL) to give the crude compound 12. b) (i) compound 12 (1.20 mmol, calculated via theory yield), nitrile (1.68 mmol), AlCl3 (1.68 mmol), CH2Cl2 (5 mL), −40 °C for 0.5 h, then at rt for 1 h under N2; (ii) ice water (1 mL); 0 °C, 5 min. Isolated yields are given.
Scheme 4: Synthesis of N(1)-unsubstituted benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepines 13. Reaction conditio...
In light of the experimental achievements and preceding theoretical work, we rationalized the synthesis with a proposed mechanism as outlined in Scheme 5 with the α-acetoxyphenylazo compound 8a serving as a model substrate. In this pathway, the reaction is initiated by AlCl3 coordination with the acetate moiety of 8a to generate the azocarbenium ion 14 as a reactive intermediate, which cannot be isolated by now [31]. Then, the nitrogen atom of the nitrile approaches to the central electron-deficient carbon atom in 14 to form a Ritter-type nitrilium salt 15 [48]. Salt 15 then undergoes a concerted but asynchronous cyclization [49] to afford the initial spiro-substituted adduct 16, which has a strong proclivity for ring expansion to occur. Accordingly, the 6-membered piperidine ring was enlarged to the 7-membered diazepine ring giving the isolated benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10 via [1,2]-shift. It is noteworthy that the intermediate products 16 bear a diazenium function where the electron-deficient N(2) displayed the feature of a latent nitrenium ion. The subsequent [1,2]-shift after cationic Huisgen-type cycloaddition occurs with complete regioselectivity to N(2) not to N(4). Meanwhile, there are two possible migrating moieties: the aromatic side competes with the aliphatic side. It has been reported that the migratory tendency of substituents prefer those with a higher ability to accommodate the respective carbocations [46]. As anticipated, it was the phenyl moiety not the aliphatic moiety that moves from C(3) to N(2) to furnish the isolated products. This rearrangement falls into the uncommon class of migration of a substituent from a carbon atom to an electron-deficient nitrogen atom [50].
![[1860-5397-14-155-i5]](/bjoc/content/inline/1860-5397-14-155-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanistic rationale for the [3+ + 2]-cycloaddition/rearrangement reaction.
Scheme 5: Mechanistic rationale for the [3+ + 2]-cycloaddition/rearrangement reaction.
The assignment of the structures for all of 5,6-dihydro-4H-benzo[b][1,2,4]triazolo[1,5-d][1,4]diazepinium salts 10 and the free base counterparts 13 was made on the basis of spectroscopic analysis. To further support our assignment, we were able to acquire X-ray crystal structures of 10k as well as of 13e, which indisputably confirm their structures [51]. For these two compounds, the ORTEP pictures are shown in Figure 2 and Figure 3, respectively.
![[1860-5397-14-155-2]](/bjoc/content/figures/1860-5397-14-155-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of salt 10k. The displacement ellipsoids are drawn at the 30% probability level.
Figure 2: Crystal structure of salt 10k. The displacement ellipsoids are drawn at the 30% probability level.
![[1860-5397-14-155-3]](/bjoc/content/figures/1860-5397-14-155-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystal structure of the free base 13e. The displacement ellipsoids are drawn at the 30% probability level.
Figure 3: Crystal structure of the free base 13e. The displacement ellipsoids are drawn at the 30% probabilit...
Conclusion
In summary, an appealing series of 1,2,4-triazole-fused 1,4-benzodiazepines 10 and 13 were prepared via the [3+ + 2]-cycloaddition reaction followed by a cationic [1,2]-rearrangement reaction. The procedure is general and has several advantages such as ready availability of starting materials, good flexibility in terms of substitution, and an unprecedented fusion pattern of the produced heterocycles. In view of the fact that the constructed 1,2,4-triazolobenzodiazepines represent a class of N-containing fused heterocycles with a new type of scaffold that is biologically interesting, the present synthetic protocol paves the way for further applications in drug-discovery research.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of NMR spectra and X-ray crystal data of compounds 10k and 13e. | ||
| Format: PDF | Size: 5.8 MB | Download |
References
-
Hadjipavlou-Litina, D.; Hansch, C. Chem. Rev. 1994, 94, 1483–1505. doi:10.1021/cr00030a002
Return to citation in text: [1] -
So, S.-S.; Karplus, M. J. Med. Chem. 1996, 39, 5246–5256. doi:10.1021/jm960536o
Return to citation in text: [1] -
Riss, J.; Cloyd, J.; Gates, J.; Collins, S. Acta Neurol. Scand. 2008, 118, 69–86. doi:10.1111/j.1600-0404.2008.01004.x
Return to citation in text: [1] -
Spencer, J.; Rathnam, R. P.; Chowdhry, B. Z. Future Med. Chem. 2010, 2, 1441–1449. doi:10.4155/fmc.10.226
Return to citation in text: [1] -
Moosmann, B.; King, L. A.; Auwärter, V. World Psychiatry 2015, 14, 248. doi:10.1002/wps.20236
Return to citation in text: [1] -
Huang, Y.; Khoury, K.; Chanas, T.; Dömling, A. Org. Lett. 2012, 14, 5916–5919. doi:10.1021/ol302837h
Return to citation in text: [1] -
Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002
Return to citation in text: [1] -
Duarte, C. D.; Barreiro, E. J.; Fraga, C. A. M. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. doi:10.2174/138955707782331722
Return to citation in text: [1] -
Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018
Return to citation in text: [1] -
Mozayani, A.; Raymon, L. P. Handbook of Drug Interactions: A Clinical and Forensic Guide, 2nd ed.; Humana Press: New York, 2011.
Return to citation in text: [1] -
Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Bioorg. Med. Chem. 2012, 20, 1878–1886. doi:10.1016/j.bmc.2011.10.080
Return to citation in text: [1] -
Bräse, S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: Cambridge, 2015.
Return to citation in text: [1] -
Sternbach, L. H. J. Med. Chem. 1979, 22, 1–7. doi:10.1021/jm00187a001
Return to citation in text: [1] -
Walser, A.; Flynn, T.; Mason, C.; Crowley, H.; Maresca, C.; Yaremko, B.; O'Donnell, M. J. Med. Chem. 1991, 34, 1209–1221. doi:10.1021/jm00107a048
Return to citation in text: [1] -
Toyo’oka, T.; Kumaki, Y.; Kanbori, M.; Kato, M.; Nakahara, Y. J. Pharm. Biomed. Anal. 2003, 30, 1773–1787. doi:10.1016/S0731-7085(02)00520-4
Return to citation in text: [1] -
Mohsin, N. A.; Qadir, M. I. Peertechz J. Med. Chem. Res. 2015, 1, 008–012. doi:10.17352/pjmcr.000002
Return to citation in text: [1] -
Boyer, S. K.; Fitchett, G.; Wasley, J. W. F.; Zaunius, G. J. Heterocycl. Chem. 1984, 21, 833–835. doi:10.1002/jhet.5570210338
Return to citation in text: [1] -
Bock, M. G.; DiPardo, R. M.; Evans, B. E.; Rittle, K. E.; Whitter, W. L.; Garsky, V. M.; Gilbert, K. F.; Leighton, J. L.; Carson, K. L.; Mellin, E. C.; Veber, D. F.; Chang, R. S. L.; Lotti, V. J.; Freedman, S. B.; Smith, A. J.; Patel, S.; Anderson, P. S.; Freidinger, R. M. J. Med. Chem. 1993, 36, 4276–4292. doi:10.1021/jm00078a018
Return to citation in text: [1] -
Wang, J.-Y.; Guo, X.-F.; Wang, D.-X.; Huang, Z.-T.; Wang, M.-X. J. Org. Chem. 2008, 73, 1979–1982. doi:10.1021/jo7024306
Return to citation in text: [1] -
Achermann, G.; Ballard, T. M.; Blasco, F.; Broutin, P.-E.; Büttelmann, B.; Fischer, H.; Graf, M.; Hernandez, M.-C.; Hilty, P.; Knoflach, F.; Koblet, A.; Knust, H.; Kurt, A.; Martin, J. R.; Masciadri, R.; Porter, R. H. P.; Stadler, H.; Thomas, A. W.; Trube, G.; Wichmann, J. Bioorg. Med. Chem. Lett. 2009, 19, 5746–5752. doi:10.1016/j.bmcl.2009.07.153
Return to citation in text: [1] -
Liu, A.; Zhou, H.; Su, G.; Zhang, W.; Yan, B. J. Comb. Chem. 2009, 11, 1083–1093. doi:10.1021/cc900109e
Return to citation in text: [1] -
Donald, J. R.; Martin, S. F. Org. Lett. 2011, 13, 852–855. doi:10.1021/ol1028404
Return to citation in text: [1] -
Zhang, G.; Smith, S. G.; Zhou, M.-M. Chem. Rev. 2015, 115, 11625–11668. doi:10.1021/acs.chemrev.5b00205
Return to citation in text: [1] -
Baud, M. G. J.; Lin-Shiao, E.; Zengerle, M.; Tallant, C.; Ciulli, A. J. Med. Chem. 2016, 59, 1492–1500. doi:10.1021/acs.jmedchem.5b01135
Return to citation in text: [1] -
Xie, H.; Liu, J.-C.; Ding, M.-W. Synthesis 2016, 48, 4541–4547. doi:10.1055/s-0036-1588308
Return to citation in text: [1] -
Khan, R.; Felix, R.; Kemmitt, P. D.; Coles, S. J.; Day, I. J.; Tizzard, G. J.; Spencer, J. Adv. Synth. Catal. 2016, 358, 98–109. doi:10.1002/adsc.201501009
Return to citation in text: [1] -
Khan, R.; Boonseng, S.; Kemmitt, P. D.; Felix, R.; Coles, S. J.; Tizzard, G. J.; Williams, G.; Simmonds, O.; Harvey, J.-L.; Atack, J.; Cox, H.; Spencer, J. Adv. Synth. Catal. 2017, 359, 3261–3269. doi:10.1002/adsc.201700626
Return to citation in text: [1] -
Křemen, F.; Gazvoda, M.; Kafka, S.; Proisl, K.; Srholcová, A.; Klásek, A.; Urankar, D.; Košmrlj, J. J. Org. Chem. 2017, 82, 715–722. doi:10.1021/acs.joc.6b01497
Return to citation in text: [1] -
Khan, R.; Felix, R.; Kemmitt, P. D.; Coles, S. J.; Tizzard, G. J.; Spencer, J. Synlett 2018, 29, 193–198. doi:10.1055/s-0036-1590920
Return to citation in text: [1] -
Vroemans, R.; Bamba, F.; Winters, J.; Thomas, J.; Jacobs, J.; Meervelt, L. V.; John, J.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 626–633. doi:10.3762/bjoc.14.49
Return to citation in text: [1] -
Wang, Q.; Jochims, J. C.; Köhlbrandt, S.; Dahlenburg, L.; Al-Talib, M.; Hamed, A.; Ismail, A. E.-H. Synthesis 1992, 710–718. doi:10.1055/s-1992-26206
Return to citation in text: [1] [2] -
Liu, X.; Liu, Y.; Wang, Q.; Zou, J. Synth. Commun. 2000, 30, 119–130. doi:10.1080/00397910008087299
Return to citation in text: [1] -
Wang, Q.; Li, Z.; Yang, H.; Li, F.; Ding, Z.; Tao, F. Synthesis 2003, 1231–1235. doi:10.1055/s-2003-39392
Return to citation in text: [1] -
Meng, Q.; Bai, H.; Li, Z.; Wang, Q.; Tao, F. Synthesis 2007, 1629–1634. doi:10.1055/s-2007-966059
Return to citation in text: [1] [2] -
Huisgen, R. Angew. Chem. 1963, 75, 604–637. doi:10.1002/ange.19630751304
Return to citation in text: [1] -
Gompper, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 312–327. doi:10.1002/anie.196903121
Return to citation in text: [1] -
Moon, M. W. J. Org. Chem. 1972, 37, 386–390. doi:10.1021/jo00968a012
Return to citation in text: [1] -
Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1973, 12, 212–224. doi:10.1002/anie.197302121
Return to citation in text: [1] -
Bradsher, C. K. Adv. Heterocycl. Chem. 1974, 16, 289–324. doi:10.1016/S0065-2725(08)60463-8
Return to citation in text: [1] -
Wei, M.-J.; Fang, D.-C.; Liu, R.-Z. Eur. J. Org. Chem. 2004, 4070–4076. doi:10.1002/ejoc.200400343
Return to citation in text: [1] -
Bercovici, D. A.; Ogilvie, J. M.; Tsvetkov, N.; Brewer, M. Angew. Chem., Int. Ed. 2013, 52, 13338–13341. doi:10.1002/anie.201306553
Return to citation in text: [1] -
Hong, X.; Bercovici, D. A.; Yang, Z.; Al-Bataineh, N.; Srinivasan, R.; Dhakal, R. C.; Houk, K. N.; Brewer, M. J. Am. Chem. Soc. 2015, 137, 9100–9107. doi:10.1021/jacs.5b04474
Return to citation in text: [1] -
Fang, Y.; Rogness, D. C.; Larock, R. C.; Shi, F. J. Org. Chem. 2012, 77, 6262–6270. doi:10.1021/jo3011073
Return to citation in text: [1] -
Lee, H.; Suzuki, M.; Cui, J.; Kozmin, S. J. Org. Chem. 2010, 75, 1756–1759. doi:10.1021/jo9025447
Return to citation in text: [1] -
Barton, D. H. R.; Jaszberenyi, J. C.; Liu, W.; Shinada, T. Tetrahedron 1996, 52, 14673–14688. doi:10.1016/0040-4020(96)00940-4
Return to citation in text: [1] -
Meng, Q.; Bai, H.; Wang, Q.; Tao, F. Synthesis 2007, 33–38. doi:10.1055/s-2006-950368
Return to citation in text: [1] [2] [3] -
Plazuk, D.; Warkentin, J.; Werstiuk, N. H. Tetrahedron 2005, 61, 5788–5796. doi:10.1016/j.tet.2005.04.024
Return to citation in text: [1] -
Cui, B.; Ren, J.; Wang, Z. J. Org. Chem. 2014, 79, 790–796. doi:10.1021/jo402383a
Return to citation in text: [1] -
Li, Z.-M.; Wang, Q.-R. Int. J. Quantum Chem. 2008, 108, 1067–1075. doi:10.1002/qua.21560
Return to citation in text: [1] -
Kroemer, R. T.; Gstach, H.; Liedl, K. R.; Rode, B. M. J. Am. Chem. Soc. 1994, 116, 6277–6283. doi:10.1021/ja00093a030
Return to citation in text: [1] -
CCDC 1438637 (10k) and CCDC 1438255 (13e) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk./data_request/cif
Return to citation in text: [1]
| 49. | Li, Z.-M.; Wang, Q.-R. Int. J. Quantum Chem. 2008, 108, 1067–1075. doi:10.1002/qua.21560 |
| 31. | Wang, Q.; Jochims, J. C.; Köhlbrandt, S.; Dahlenburg, L.; Al-Talib, M.; Hamed, A.; Ismail, A. E.-H. Synthesis 1992, 710–718. doi:10.1055/s-1992-26206 |
| 48. | Cui, B.; Ren, J.; Wang, Z. J. Org. Chem. 2014, 79, 790–796. doi:10.1021/jo402383a |
| 1. | Hadjipavlou-Litina, D.; Hansch, C. Chem. Rev. 1994, 94, 1483–1505. doi:10.1021/cr00030a002 |
| 2. | So, S.-S.; Karplus, M. J. Med. Chem. 1996, 39, 5246–5256. doi:10.1021/jm960536o |
| 3. | Riss, J.; Cloyd, J.; Gates, J.; Collins, S. Acta Neurol. Scand. 2008, 118, 69–86. doi:10.1111/j.1600-0404.2008.01004.x |
| 4. | Spencer, J.; Rathnam, R. P.; Chowdhry, B. Z. Future Med. Chem. 2010, 2, 1441–1449. doi:10.4155/fmc.10.226 |
| 5. | Moosmann, B.; King, L. A.; Auwärter, V. World Psychiatry 2015, 14, 248. doi:10.1002/wps.20236 |
| 15. | Toyo’oka, T.; Kumaki, Y.; Kanbori, M.; Kato, M.; Nakahara, Y. J. Pharm. Biomed. Anal. 2003, 30, 1773–1787. doi:10.1016/S0731-7085(02)00520-4 |
| 46. | Meng, Q.; Bai, H.; Wang, Q.; Tao, F. Synthesis 2007, 33–38. doi:10.1055/s-2006-950368 |
| 13. | Sternbach, L. H. J. Med. Chem. 1979, 22, 1–7. doi:10.1021/jm00187a001 |
| 14. | Walser, A.; Flynn, T.; Mason, C.; Crowley, H.; Maresca, C.; Yaremko, B.; O'Donnell, M. J. Med. Chem. 1991, 34, 1209–1221. doi:10.1021/jm00107a048 |
| 47. | Plazuk, D.; Warkentin, J.; Werstiuk, N. H. Tetrahedron 2005, 61, 5788–5796. doi:10.1016/j.tet.2005.04.024 |
| 7. | Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002 |
| 8. | Duarte, C. D.; Barreiro, E. J.; Fraga, C. A. M. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. doi:10.2174/138955707782331722 |
| 9. | Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018 |
| 10. | Mozayani, A.; Raymon, L. P. Handbook of Drug Interactions: A Clinical and Forensic Guide, 2nd ed.; Humana Press: New York, 2011. |
| 11. | Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Bioorg. Med. Chem. 2012, 20, 1878–1886. doi:10.1016/j.bmc.2011.10.080 |
| 12. | Bräse, S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: Cambridge, 2015. |
| 46. | Meng, Q.; Bai, H.; Wang, Q.; Tao, F. Synthesis 2007, 33–38. doi:10.1055/s-2006-950368 |
| 6. | Huang, Y.; Khoury, K.; Chanas, T.; Dömling, A. Org. Lett. 2012, 14, 5916–5919. doi:10.1021/ol302837h |
| 34. | Meng, Q.; Bai, H.; Li, Z.; Wang, Q.; Tao, F. Synthesis 2007, 1629–1634. doi:10.1055/s-2007-966059 |
| 35. | Huisgen, R. Angew. Chem. 1963, 75, 604–637. doi:10.1002/ange.19630751304 |
| 36. | Gompper, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 312–327. doi:10.1002/anie.196903121 |
| 37. | Moon, M. W. J. Org. Chem. 1972, 37, 386–390. doi:10.1021/jo00968a012 |
| 38. | Schmidt, R. R. Angew. Chem., Int. Ed. Engl. 1973, 12, 212–224. doi:10.1002/anie.197302121 |
| 39. | Bradsher, C. K. Adv. Heterocycl. Chem. 1974, 16, 289–324. doi:10.1016/S0065-2725(08)60463-8 |
| 43. | Fang, Y.; Rogness, D. C.; Larock, R. C.; Shi, F. J. Org. Chem. 2012, 77, 6262–6270. doi:10.1021/jo3011073 |
| 44. | Lee, H.; Suzuki, M.; Cui, J.; Kozmin, S. J. Org. Chem. 2010, 75, 1756–1759. doi:10.1021/jo9025447 |
| 51. | CCDC 1438637 (10k) and CCDC 1438255 (13e) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk./data_request/cif |
| 31. | Wang, Q.; Jochims, J. C.; Köhlbrandt, S.; Dahlenburg, L.; Al-Talib, M.; Hamed, A.; Ismail, A. E.-H. Synthesis 1992, 710–718. doi:10.1055/s-1992-26206 |
| 32. | Liu, X.; Liu, Y.; Wang, Q.; Zou, J. Synth. Commun. 2000, 30, 119–130. doi:10.1080/00397910008087299 |
| 33. | Wang, Q.; Li, Z.; Yang, H.; Li, F.; Ding, Z.; Tao, F. Synthesis 2003, 1231–1235. doi:10.1055/s-2003-39392 |
| 34. | Meng, Q.; Bai, H.; Li, Z.; Wang, Q.; Tao, F. Synthesis 2007, 1629–1634. doi:10.1055/s-2007-966059 |
| 45. | Barton, D. H. R.; Jaszberenyi, J. C.; Liu, W.; Shinada, T. Tetrahedron 1996, 52, 14673–14688. doi:10.1016/0040-4020(96)00940-4 |
| 17. | Boyer, S. K.; Fitchett, G.; Wasley, J. W. F.; Zaunius, G. J. Heterocycl. Chem. 1984, 21, 833–835. doi:10.1002/jhet.5570210338 |
| 18. | Bock, M. G.; DiPardo, R. M.; Evans, B. E.; Rittle, K. E.; Whitter, W. L.; Garsky, V. M.; Gilbert, K. F.; Leighton, J. L.; Carson, K. L.; Mellin, E. C.; Veber, D. F.; Chang, R. S. L.; Lotti, V. J.; Freedman, S. B.; Smith, A. J.; Patel, S.; Anderson, P. S.; Freidinger, R. M. J. Med. Chem. 1993, 36, 4276–4292. doi:10.1021/jm00078a018 |
| 19. | Wang, J.-Y.; Guo, X.-F.; Wang, D.-X.; Huang, Z.-T.; Wang, M.-X. J. Org. Chem. 2008, 73, 1979–1982. doi:10.1021/jo7024306 |
| 20. | Achermann, G.; Ballard, T. M.; Blasco, F.; Broutin, P.-E.; Büttelmann, B.; Fischer, H.; Graf, M.; Hernandez, M.-C.; Hilty, P.; Knoflach, F.; Koblet, A.; Knust, H.; Kurt, A.; Martin, J. R.; Masciadri, R.; Porter, R. H. P.; Stadler, H.; Thomas, A. W.; Trube, G.; Wichmann, J. Bioorg. Med. Chem. Lett. 2009, 19, 5746–5752. doi:10.1016/j.bmcl.2009.07.153 |
| 21. | Liu, A.; Zhou, H.; Su, G.; Zhang, W.; Yan, B. J. Comb. Chem. 2009, 11, 1083–1093. doi:10.1021/cc900109e |
| 22. | Donald, J. R.; Martin, S. F. Org. Lett. 2011, 13, 852–855. doi:10.1021/ol1028404 |
| 23. | Zhang, G.; Smith, S. G.; Zhou, M.-M. Chem. Rev. 2015, 115, 11625–11668. doi:10.1021/acs.chemrev.5b00205 |
| 24. | Baud, M. G. J.; Lin-Shiao, E.; Zengerle, M.; Tallant, C.; Ciulli, A. J. Med. Chem. 2016, 59, 1492–1500. doi:10.1021/acs.jmedchem.5b01135 |
| 25. | Xie, H.; Liu, J.-C.; Ding, M.-W. Synthesis 2016, 48, 4541–4547. doi:10.1055/s-0036-1588308 |
| 26. | Khan, R.; Felix, R.; Kemmitt, P. D.; Coles, S. J.; Day, I. J.; Tizzard, G. J.; Spencer, J. Adv. Synth. Catal. 2016, 358, 98–109. doi:10.1002/adsc.201501009 |
| 27. | Khan, R.; Boonseng, S.; Kemmitt, P. D.; Felix, R.; Coles, S. J.; Tizzard, G. J.; Williams, G.; Simmonds, O.; Harvey, J.-L.; Atack, J.; Cox, H.; Spencer, J. Adv. Synth. Catal. 2017, 359, 3261–3269. doi:10.1002/adsc.201700626 |
| 28. | Křemen, F.; Gazvoda, M.; Kafka, S.; Proisl, K.; Srholcová, A.; Klásek, A.; Urankar, D.; Košmrlj, J. J. Org. Chem. 2017, 82, 715–722. doi:10.1021/acs.joc.6b01497 |
| 29. | Khan, R.; Felix, R.; Kemmitt, P. D.; Coles, S. J.; Tizzard, G. J.; Spencer, J. Synlett 2018, 29, 193–198. doi:10.1055/s-0036-1590920 |
| 30. | Vroemans, R.; Bamba, F.; Winters, J.; Thomas, J.; Jacobs, J.; Meervelt, L. V.; John, J.; Dehaen, W. Beilstein J. Org. Chem. 2018, 14, 626–633. doi:10.3762/bjoc.14.49 |
| 46. | Meng, Q.; Bai, H.; Wang, Q.; Tao, F. Synthesis 2007, 33–38. doi:10.1055/s-2006-950368 |
| 16. | Mohsin, N. A.; Qadir, M. I. Peertechz J. Med. Chem. Res. 2015, 1, 008–012. doi:10.17352/pjmcr.000002 |
| 40. | Wei, M.-J.; Fang, D.-C.; Liu, R.-Z. Eur. J. Org. Chem. 2004, 4070–4076. doi:10.1002/ejoc.200400343 |
| 41. | Bercovici, D. A.; Ogilvie, J. M.; Tsvetkov, N.; Brewer, M. Angew. Chem., Int. Ed. 2013, 52, 13338–13341. doi:10.1002/anie.201306553 |
| 42. | Hong, X.; Bercovici, D. A.; Yang, Z.; Al-Bataineh, N.; Srinivasan, R.; Dhakal, R. C.; Houk, K. N.; Brewer, M. J. Am. Chem. Soc. 2015, 137, 9100–9107. doi:10.1021/jacs.5b04474 |
| 50. | Kroemer, R. T.; Gstach, H.; Liedl, K. R.; Rode, B. M. J. Am. Chem. Soc. 1994, 116, 6277–6283. doi:10.1021/ja00093a030 |
© 2018 Luan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)