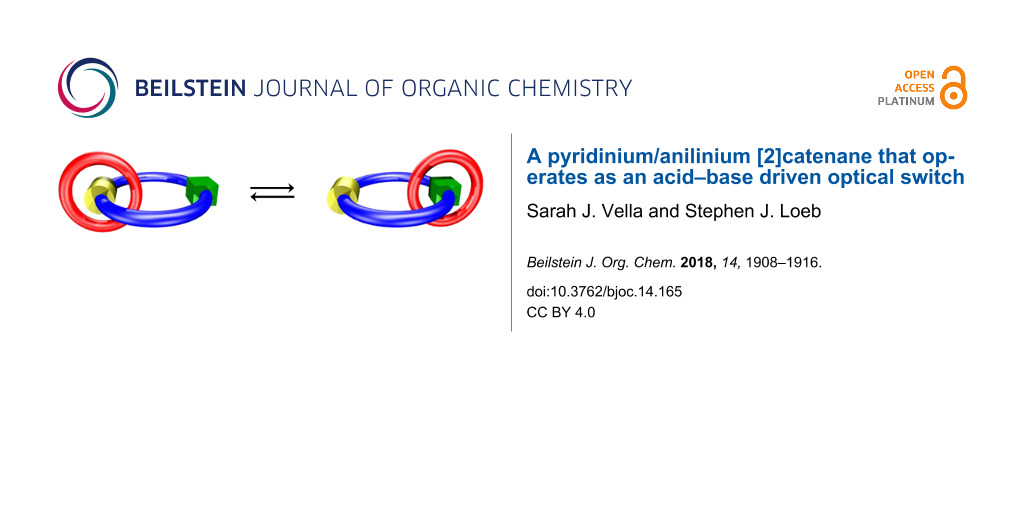
Abstract
A two-station [2]catenane containing a large macrocycle with two different recognition sites, one bis(pyridinium)ethane and one benzylanilinium, as well as a smaller DB24C8 ring was synthesized and characterized. 1H NMR spectroscopy showed that the DB24C8 ring can shuttle between the two recognition sites depending on the protonation state of the larger macrocycle. When the aniline group is neutral, the DB24C8 ring resides solely at the bis(pyridinium)ethane site, while addition of acid forms a charged benzylanilinium site. The DB24C8 then shuttles between the two charged recognition sites with occupancy favoring the bis(pyridinium)ethane site by a ratio of 4:1. The unprotonated [2]catenane has a deep yellow/orange color when the DB24C8 ring resides solely at the bis(pyridinium)ethane site and changes to colorless when the crown ether is shuttling (i.e., circumrotating) back and forth between the two recognition sites thus optically signalling the onset of the shuttling dynamics.

Graphical Abstract
Introduction
[2]Rotaxane molecular shuttles [1-5] are the dynamic building blocks of a wide variety of molecular switches [6-9] and a number of sophisticated molecular machines that operate away from equilibrium [10-15]. We have previously reported [2]rotaxane molecular switches containing a single dibenzo[24]crown ether DB24C8 wheel and two different recognition sites; benzylanilinium and 1,2-bis(pyridinium)ethane [16]. These shuttles operate as bistable switches driven by acid/base chemistry and can be optically sensed by either a change in color (yellow/colorless) for [1F![[Graphic 2]](/bjoc/content/inline/1860-5397-14-165-i3.svg?max-width=637&scale=0.945456) DB24C8]2+ or a fluorescence change (OFF/ON) for [1A
DB24C8]2+ or a fluorescence change (OFF/ON) for [1A![[Graphic 1]](/bjoc/content/inline/1860-5397-14-165-i2.svg?max-width=637&scale=0.945456) DB24C8]2+; see Figure 1.
DB24C8]2+; see Figure 1.
![[1860-5397-14-165-1]](/bjoc/content/figures/1860-5397-14-165-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Two [2]rotaxane molecular shuttles with both bis(pyridinium)ethane and benzylanilinium recognition sites that can be switched by acid–base chemistry and optically sensed by a) a color change from colorless to yellow and b) a change in fluorescence from OFF to ON (CD3CN or CD2Cl2). Code: F to indicate CF3 groups; A to indicate anthracene group.
Figure 1: Two [2]rotaxane molecular shuttles with both bis(pyridinium)ethane and benzylanilinium recognition ...
In addition, we have also previously prepared a [3]catenane containing two dibenzo[24]crown ether DB24C8 rings interlocked onto a much larger macrocyclic ring containing two 1,2-bis(pyridinium)ethane recognition sites linked by terphenyl spacer groups [17] (Figure 2).
![[1860-5397-14-165-2]](/bjoc/content/figures/1860-5397-14-165-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A [3]catenane containing two identical bis(pyridinium)ethane recognition sites on a large macrocycle and two smaller threaded DB24C8 rings.
Figure 2: A [3]catenane containing two identical bis(pyridinium)ethane recognition sites on a large macrocycl...
It was thus of interest to design and build these two different recognition sites (benzylanilinium and bis(pyridinium)ethane) into an analogous circumrotational [2]catenane molecular switch to compare to the linear [2]rotaxane molecular shuttles outlined in Figure 1. This should be possible because of the structural similarities (size and shape) between the bis(pyridinium)ethane and benzylanilinium recognition sites. Each has a two-atom chain in a low energy, anti-conformation linking aromatic rings and the distance between the terminal nitrogen atoms are 18.11 and 18.09 Å (MM3) for the benzylaniline and bis(dipyridinium)ethane axles 4 and 52+, respectively; see Figure 2 and Scheme 1 compound [8DB24C8]6+ for this comparison and concept.
Results and Discussion
Synthesis
Although the previously reported [3]catenane (Figure 2) was synthesized using a one-step, self-assembly procedure from two bis(pyridinium)ethane axles, two terphenyl spacers and two DB24C8 crown ethers, a [2]catenane with different recognition sites requires a stepwise approach involving the incorporation of each recognition site independently. Overall, the synthesis of [2]catenane [8DB24C8]6+ required multiple steps and is outlined in Scheme 1. Two literature preparations were used to construct each of the known compounds, terphenyl linker 6 [18] and bis(pyridinium)ethane axle [5][OTf]2 [19,20], while the new benzylaniline axle 4 was prepared as shown from 3 [21]. Once the precursor components were synthesized, the [2]catenane was assembled in two steps. Firstly, [5][OTf]2 was reacted with ten equivalents of the bis(bromomethyl)terphenyl linker 6 in CH3CN to afford [7][OTf]4 in moderate yield. Secondly, the [2]pseudorotaxane [7DB24C8]4+ was formed using [7][OTf]4 in the presence of DB24C8 followed by ring closure using the benzylaniline axle 4 to yield [8DB24C8][OTf]6. The reaction was performed under dilute conditions with 10 equivalents of crown ether to favor ring closure and kinetic trapping of the smaller ring.
![[1860-5397-14-165-i1]](/bjoc/content/inline/1860-5397-14-165-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1:
Step-wise synthesis of [2]catenane [8![[Graphic 3]](/bjoc/content/inline/1860-5397-14-165-i4.svg?max-width=637&scale=0.827274) DB24C8]6+ containing benzylanilinium and bis(pyridinium)ethane recognition sites and terphenyl spacers.
DB24C8]6+ containing benzylanilinium and bis(pyridinium)ethane recognition sites and terphenyl spacers.
Scheme 1:
Step-wise synthesis of [2]catenane [8DB24C8]6+ containing benzylanilinium and bis(pyridinium)ethane...
To isolate the pure [2]catenane, the reaction solvent (CH3CN) was evaporated and the residue washed with toluene to remove excess crown ether. This was then followed by column chromatography on silica gel using a 5:3:2 mixture of CH3OH/2 M NH4Cl/CH3NO2 as the eluent. Fractions containing the product (Rf = 0.66) were combined and anion exchanged to the triflate salt to yield [2]catenane [8DB24C8][OTf]6.
Characterization
The 1H NMR spectrum of [2]catenane [8DB24C8]6+ (298 K, CD2Cl2) is shown in Figure 3 and the labelling scheme for the H-atoms is given in Scheme 1. All resonances were assigned based on 2D COSY NMR spectroscopy as well as comparison to 1H NMR and COSY spectra of individual components 6 and 74+. Comparing the proton chemicals shifts for H-atoms n–y of [8DB24C8]6+ with those of precursor 74+ shows changes in chemical shift typically associated with the close interaction of DB24C8 with a bis(pyridinium)ethane recognition site [18]. In particular, the significant downfield shifts observed for ethylene protons s and t from 5.30 ppm in 74+ to 5.56 ppm for [8DB24C8]6+ as well as u and r, the ortho pyridinium protons, from 9.04 ppm in 74+ to 9.31 ppm for [8DB24C8]6+ are characteristic of hydrogen-bonding to the crown ether. In addition, π-stacking interactions induce upfield shifts for protons p, q, v and w from 8.48 ppm in 74+ to 8.24 ppm for [8DB24C8]6+. Protons o, x, n and y do not shift appreciably because the crown ether does not extend far enough to interact with these protons. In contrast, the chemical shifts for protons a–d and I–L on the benzylaniline portion of the large ring of [8DB24C8]6+ do not shift significantly inferring that in the neutral aniline state the crown ether resides exclusively at the bis(pyridinium)ethane site of the [2]catenane. Table 1 summarizes the chemical shift differences between the [2]catenane [8DB24C8]6+ and precursor 74+ which contains no crown ether.
![[1860-5397-14-165-3]](/bjoc/content/figures/1860-5397-14-165-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3:
1H NMR spectrum of [2]catenane [8DB24C8]6+ (500 MHz, 298 K, CD2Cl2) showing the assigned proton chemical shifts; see Scheme 1 for labelling.
Figure 3:
1H NMR spectrum of [2]catenane [8DB24C8]6+ (500 MHz, 298 K, CD2Cl2) showing the assigned proton che...
Table 1:
Summary of major chemical shift differences between precursor 74+ and catenane [8DB24C8]6+.
| protonsa | 74+ |
[8DB24C8]6+
|
|---|---|---|
| n, y | 5.89 | 5.90 |
| o, x | 9.05 | 9.03 |
| p, w | 8.47 | 8.19 |
| q, v | 8.50 | 8.22 |
| r, u | 9.04 | 9.31 |
| s, t | 5.30 | 5.56 |
aAll chemical shift values given in ppm relative to TMS in CD3CN at 298 K.
A sample of [8DB24C8]6+ (1:1 CH3OH/CH3CN) was analyzed by high-resolution electrospray mass spectrometry (HRESIMS). Sufficient resolution for each of the 2+, 3+, 4+ and 5+ molecular ions allowed for exact mass measurements (<5 ppm) confirming the catenated nature of the structure. Table 2 summarizes the observed values.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-165-i4.svg?max-width=637&scale=0.7)
Acid–base driven switching
The analysis of the 1H NMR spectrum (CD3CN, 298 K) of [8DB24C8]6+ indicates that the DB24C8 ring resides exclusively at the bis(pyridinium)ethane recognition site. This is easily understood as the neutral benzylaniline site does not allow for appreciable non-covalent interactions and cannot compete for the DB24C8 ring with the dicationic bis(pyridinium)ethane site. However, the addition of one equivalent of triflic acid (CF3SO3H) to a solution of [8DB24C8]6+ results in protonation of the aniline nitrogen atom to give [8-HDB24C8]7+ and a second viable recognition site for the crown ether.
Figure 4 shows a partial 1H NMR spectrum of protonated [8-HDB24C8]7+ in CD3CN at 298 K. The smaller DB24C8 ring can now reside at either of the bis(pyridinium)ethane or benzylanilinium sites and these two possible co-conformations are designated A and B in Figure 4a. The ethylene protons at the core of the bis(pyridinium)ethane motif, labelled s and t in A and s’ and t’ in B are clearly distinguishable and show that there is a 4:1 ratio of A:B indicating that the smaller DB24C8 ring prefers to occupy the bis(pyridinium)ethane site over the benzylanilinium site and that shuttling between the two sites is slow on the NMR timescale under these experimental conditions. Addition of base (NEt3) returns the system to its original state and the process can be cycled by repeated addition of acid (CF3SO3H) and base without significant degradation of the compound as verified by 1H NMR spectroscopy.
![[1860-5397-14-165-4]](/bjoc/content/figures/1860-5397-14-165-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4:
a) The [2]catenane [8DB24C8]6+ can be protonated to yield [8-HDB24C8]7+ in two different co-conformations A and B. b) The partial 1H NMR spectrum (500 MHz, 298 K, CD3CN) of [8-HDB24C8]7+ shows key resonances for both co-conformations A (red) and B (blue). See Scheme 1 for labelling; atoms of co-conformer B are labelled with a prime, e.g., s’ versus s.
Figure 4:
a) The [2]catenane [8DB24C8]6+ can be protonated to yield [8-HDB24C8]7+ in two different co-conform...
Interestingly, these results are contrary to those observed for the [2]rotaxane molecular shuttles [1FDB24C8]2+ and [1ADB24C8]2+ shown in Figure 1. For that system, the benzylanilinium site was preferred 3:1 for [1FDB24C8]2+ and 9:1 for [1ADB24C8]2+ in CD3CN and when CD2Cl2 was used the systems were completely bistable with DB24C8 preferring to reside exclusively at the bis(pyridinium)ethane site when unprotonated and exclusively at the benzylanilinium site when protonated.
The UV–visible spectra of [8DB24C8]6+ and [8-HDB24C8]7+ are shown in Figure 5 for 2.0 × 10−5 M solutions in CH3CN. The molar absorptivity (ε) of [8DB24C8]6+ was calculated to be 22,680 L mol−1 cm−1 with λmax at 412 nm. The large absorption is due to an intramolecular charge transfer (ICT) band arising from charge transfer between the aniline nitrogen and pyridinium group of the benzylanilinium recognition site. However, this ICT band (412 nm) is eliminated by protonating the aniline nitrogen to form [8-HDB24C8]7+. Therefore, when the [2]catenane absorbs strongly showing a deep yellow/orange solution this indicates that the crown resides solely on the bis(pyridinium)ethane site for [8DB24C8]6+ but, when the [2]catenane does not absorb in the UV–visible region yielding a colorless solution this means the crown ether must be shuttling (i.e., circumrotating) back and forth between the two co-conformations, A and B, of [8-HDB24C8]7+.
![[1860-5397-14-165-5]](/bjoc/content/figures/1860-5397-14-165-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5:
UV–visible spectra of [8DB24C8]6+ (orange trace) and [8-HDB24C8]7+ (black trace) in CH3CN solution at 2.0 × 10−5 M and 298 K.
Figure 5:
UV–visible spectra of [8DB24C8]6+ (orange trace) and [8-HDB24C8]7+ (black trace) in CH3CN solution ...
Conclusion
A two-station circumrotational [2]catenane has been synthesized and its operation described. The system consists of a large macrocycle containing two different recognition sites, one bis(pyridinium)ethane and one benzylanilinium with a single smaller DB24C8 ring that can shuttle between the two recognition sites depending on the protonation state of the larger macrocycle. When the aniline group is neutral, the DB24C8 ring resides only at the bis(pyridinium)ethane site. However, addition of acid activates the benzylanilinium site allowing the ring to shuttle between the two, now competing, recognition sites. It was found that DB24C8 prefers the bis(pyridinium)ethane site over the protonated benzylanilinium site in a ratio of 4:1. This is quite different from similar [2]rotaxane molecular shuttles (Figure 1) where, once protonated, the benzylanilinium site was preferred (CD3CN) and in some cases exclusively (CD2Cl2) generating a true ON/OFF bistable switch; unfortunately, the [2]catenane switch is insoluble in CD2Cl2 when protonated so a comparison could not be undertaken in this solvent. This difference in site populations between [2]rotaxane and [2]catenane is due to the presence of electron-withdrawing CF3 groups on the [2]rotaxane which make the benzylanilinium site more favorable in this case. Since it is fairly straightforward to change the nature of the stoppering groups of a [2]rotaxane dumbbell while the cyclic nature of the large ring makes it difficult to derivatize, [2]rotaxanes are deemed easier to fine-tune from a structural perspective than [2]catenanes. Although we were able to create an optically sensitive [2]catenane molecular shuttle with the bis(pyridinium)ethane and benzylanilinium recognition motifs, we could not achieve the true ON/OFF, bistable molecular switching previously observed for analogous [2]rotaxanes.
Experimental
General comments
4-Bromobenzyl bromide, 4-bromoaniline, 4-pyridylboronic acid, 1,3-dichlorobenzene, p-tolylmagnesium bromide, n-butyllithium and N-bromosuccinimide were purchased from Aldrich and used as received. Benzoyl peroxide was purchased from Acros and used as received. Compounds 3 [18], [5][OTf]2 [19,20] and 6 [21] were prepared using literature methods. Solvents were dried using an Innovative Technologies solvent purification system. Thin-layer chromatography (TLC) was performed using Teledyne Silica gel 60 F254 plates and viewed under UV light. Column chromatography was performed using Silicycle ultra-pure silica gel (230–400 mesh). The solvents were dried and distilled prior to use. NMR spectra were recorded on a Bruker Avance III console equipped with an 11.7 T magnet (e.g., 500 MHz for 1H). Samples were locked to the deuterated solvent and all chemical shifts reported in ppm referenced to tetramethylsilane. Mass spectra were recorded on a Waters Xevo G2-XS instrument. Solutions with concentrations of 0.001 molar were prepared in methanol and injected for analysis at a rate of 5 µL/min using a syringe pump.
Synthesis of 4
DMF (250 mL) and H2O (100 mL) were added to a round bottom Schlenk flask (500 mL) and degassed with N2 for 2 h. To this solvent mixture, 3 (1.11 g, 0.00325 mol), 4-pyridylboronic acid (1.00 g, 0.00814 mol) and Na2CO3 (2.07 g, 0.195 mol) were added and the solution degassed for an additional 1 h. Catalyst [Pd(PPh3)4] (0.188 g, 16.3 mmol) was added and the solution degassed for an additional 30 min. The reaction was then refluxed for 5 days and the progress of the reaction monitored using 1H NMR spectroscopy. After the 5 days, the reaction was cooled to room temperature and the solvents removed by evaporation. The residue was dissolved in CH2Cl2 (100 mL) and washed with H2O (3 × 50 mL). The CH2Cl2 layer was dried over anhydrous MgSO4, filtered and concentrated. Compound 4 precipitated as a pale yellow powder which was collected by vacuum filtration. The filtrate was then evaporated under vacuum and the residue subjected to column chromatography (SiO2, 1% MeOH/CHCl3, Rf = 0.13) to yield further product. The batches of product (from precipitate and filtrate) were combined and recrystallized from acetone. Yield, 0.800 g, 73%; mp 186–188 °C; 1H NMR (500 MHz, CD3CN, 298 K) δ 8.60 (d, 3J = 6.1 Hz, 2H), 8.48 (d, 3J = 6.1 Hz, 2H), 7.71 (d, 3J = 8.2 Hz, 2H), 7.59 (d, 3J = 6.2 Hz, 2H), 7.54 (d, 3J = 8.7 Hz, 2H), 7.51 (d, 3J = 8.2 Hz, 2H), 7.49 (d, 3J = 6.2 Hz, 2H), 6.73 (d, 3J = 8.7 Hz, 2H), 5.41 (br t, 1H), 4.46 (d, 3J = 6.2 Hz, 2H); 13C NMR (125 MHz, CD3CN, 298 K) δ 151.1, 150.3, 149.8, 147.6, 146.8, 140.5, 140.2, 129.9, 128.4, 127.9, 127.6, 120.8, 120.4, 113.1, 47.6; HRMS (ESI) m/z: [M + H]+ calcd for [C23H20N3]+, 338.1657; found, 338.1650.
Synthesis of [7][OTf]4
[5][OTf]2 (0.400 g, 0.626 mmol) and 6 (2.61 g, 6.26 mmol) were dissolved in CH3CN (75 mL) and stirred at room temperature for 7 days. The resulting precipitate was filtered, collected and stirred in CH2Cl2 for 20 min and filtered to remove excess 6. The precipitate was then anion exchanged to the triflate salt in a two-layer CH3NO2/NaOTf(aq) solution. The layers were separated and the CH3NO2 layer washed with H2O (3 × 5 mL) and then dried over anhydrous MgSO4. The CH3NO2 was removed by rotary evaporation and [7][OTf]4 isolated as a pale yellow solid. Yield 0.700 g, 69%; mp >180 °C (dec.); 1H NMR (500 MHz, CD3CN, 298 K) δ 9.05 (d, 3J = 6.9 Hz, 4H), 9.04 (d, 3J = 6.9 Hz, 4H), 8.50 (d, 3J = 6.1 Hz, 4H), 8.47 (d, 3J = 6.1 Hz, 4H), 7.91 (s, 2H), 7.86 (d, 3J = 8.2 Hz, 4H), 7.71 (d, 3J = 8.2 Hz, 4H), 7.68 (d, 3J = 7.9 Hz, 2H), 7.67 (d, 3J = 8.2 Hz, 4H), 7.62 (d, 3J = 8.0 Hz, 4H), 7.57 (t, 3J = 7.9, 3J = 8.1 Hz, 2H), 7.54 (d, 3J = 8.2 Hz, 4H), 5.89 (s, 4H), 5.30 (br s, 4H), 4.66 (s, 4H); HRMS (ESI) m/z: [M − OTf]+ calcd, 1457.1114; found, 1457.1144.
Synthesis of [8DB24C8][OTf]6
[7][OTf]4 (0.155 g, 0.0963 mmol) and DB24C8 (0.432 g, 0.963 mmol) were dissolved in a two phase CH3NO2/H2O mixture and stirred at room temperature for 30 min to allow [2]pseudorotaxane formation. Compound 4 (0.0330 g, 0.0963 mmol) was then added along with NaOTf (0.0330 g, 0.193 mmol) and the reaction stirred at room temperature for 21 days. The water layer was separated and the CH3NO2 evaporated. The resulting residue was washed with CH2Cl2 (3 × 10 mL) to remove excess DB24C8 and subjected to column chromatography on silica gel (5:3:2 mixture of CH3OH/NH4Cl (2 M)/CH3NO2). Fractions containing the product (Rf = 0.66) were combined and the solvents evaporated. The residue was dissolved in a two layer CH3NO2/NaOTf(aq) solution to anion exchange to the triflate salt. The H2O layer was removed and the CH3NO2 layer washed with H2O (3 × 5 mL) to extract any remaining salts. The CH3NO2 layer was dried with anhydrous MgSO4 and then evaporated to yield [8DB24C8][OTf]6 as a yellow-orange solid. Yield 0.030 g, 12%; mp >210 °C (dec.); HRMS (ESI) m/z: [M − 2OTf]2+ calcd for [C113H103F12N7O20S4]2+, 1116.7969, found, 1116.7972; [M − 3OTf]3+ calcd for [C112H103F9N7O17S3]3+, 694.8804, found, 694.8835; [M − 4OTf]4+ calcd for [C111H103F6N7O14S2]4+, 483.9222, found, 483.9246; [M − 5OTf]5+ calcd for [C110H103F3N7O11S]5+, 357.3472, found, 357.3465; 1H NMR (500 MHz, CD2Cl2, 298 K) δ 9.31 (d, 3Jrq = 6.7 Hz, 2H, r), 9.31 (d, 3Juv = 6.7 Hz, 2H, u), 9.03 (d, 3Jop = 6.8 Hz, 2H, o), 9.03 (d, 3Jxw = 6.8 Hz, 2H, x), 8.76 (d, 3JIJ = 6.8 Hz, 2H, I), 8.49 (d, 3Jdc = 6.9 Hz, 2H, d), 8.22 (d, 3Jqr = 6.7 Hz, 2H, q), 8.22 (d, 3Jvu = 6.7 Hz, 2H, v), 8.22 (d, 3JJI = 6.8 Hz, 2H, J), 8.19 (d, 3Jpo = 6.8 Hz, 2H, p), 8.19 (d, 3Jwx = 6.8 Hz, 2H, w), 8.04 (d, 3Jcd = 6.9 Hz, 2H, c), 7.87 (d, 3Jlm = 8.2 Hz, 2H, l), 7.87 (d, 3JAz = 8.2 Hz, 2H, A), 7.87 (d, 3JKL = 8.2 Hz, 2H, K), 7.83 (s, 1H, k), 7.83 (s, 1H, E), 7.80 (d, 3Jgf = 8.4 Hz, 2H, g), 7.78 (d, 3Jba = 8.7 Hz, 2H, b), 7.77 (d, 3JFG = 8.6 Hz, 2H, F), 7.74–7.70 (d, 1H, h), 7.74–7.70 (d, 1H, j), 7.74–7.70 (d, 1H, B), 7.74–7.70 (d, 1H, D), 7.64 (d, 3Jml = 8.2 Hz, 2H, m), 7.64 (d, 3JzA = 8.2 Hz, 2H, z), 7.62 (dd, 1H, i), 97.62 (dd, 1H, C), 7.58 (d, 3JLK = 8.2 Hz, 2H, L), 7.55 (d, 3JGF = 8.6 Hz, 2H, G), 7.51 (d, 3Jfg = 8.4 Hz, 2H, f), 6.79 (d, 3Jab = 8.7 Hz, 2H, a), 6.62 (m, 3Jortho = 5.8, 3Jmeta = 3.6 Hz, 4H, 1), 6.33 (m, 3Jortho = 5.8, 3Jmeta = 3.6 Hz, 4H, 2), 5.90 (s, 2H, n), 5.90 (s, 2H, y), 5.87 (t, 3JNM = 5.6 Hz, 1H, N), 5.74 (d, 2H, H), 5.59 (s, 2H, e), 5.56 (s, 2H, s), 5.56 (s, 2H, t), 4.48 (d, 3JMN = 5.6 Hz, 2H, M), 4.04–3.99 (m, 24H, 3–5).
Acknowledgements
The authors thank NSERC of Canada for funding; SJL for a Discovery grant and SJV for a post graduate scholarship. The authors acknowledge that the majority of this material was sourced from Vella, S. J. New Interlocked Molecular Machines. Ph.D. Thesis, University of Windsor, Windsor, ON, Canada, 2006.
References
-
Bruns, C. J.; Stoddart, J. F. The Nature of the Mechanical Bond; John Wiley & Sons: Hoboken, New Jersey, 2017.
Return to citation in text: [1] -
Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216
Return to citation in text: [1] -
Anelli, P. L.; Spencer, N.; Stoddart, J. F. J. Am. Chem. Soc. 1991, 113, 5131–5133. doi:10.1021/ja00013a096
Return to citation in text: [1] -
Zhu, K.; Vukotic, V. N.; Loeb, S. J. Angew. Chem., Int. Ed. 2012, 51, 2168–2172. doi:10.1002/anie.201108488
Return to citation in text: [1] -
Vukotic, V. N.; Zhu, K.; Baggi, G.; Loeb, S. J. Angew. Chem., Int. Ed. 2017, 56, 6136–6141. doi:10.1002/anie.201612549
Return to citation in text: [1] -
Bruns, C. J.; Stoddart, J. F. Acc. Chem. Res. 2014, 47, 2186–2199. doi:10.1021/ar500138u
Return to citation in text: [1] -
Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791
Return to citation in text: [1] -
Thordarson, P.; Bijsterveld, E. J. A.; Rowan, A. E.; Nolte, R. J. M. Nature 2003, 424, 915–918. doi:10.1038/nature01925
Return to citation in text: [1] -
Xue, M.; Yang, Y.; Chi, X.; Yan, X.; Huang, F. Chem. Rev. 2015, 115, 7398–7501. doi:10.1021/cr5005869
Return to citation in text: [1] -
Ragazzon, G.; Baroncini, M.; Silvi, S.; Venturi, M.; Credi, A. Nat. Nanotechnol. 2015, 10, 70–75. doi:10.1038/nnano.2014.260
Return to citation in text: [1] -
Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455
Return to citation in text: [1] -
Collier, C. P.; Mattersteig, G.; Wong, E. W.; Luo, Y.; Beverly, K.; Sampaio, J.; Raymo, F. M.; Stoddart, J. F.; Heath, J. R. Science 2000, 289, 1172–1175. doi:10.1126/science.289.5482.1172
Return to citation in text: [1] -
Fahrenbach, A. C.; Warren, S. C.; Incorvati, J. T.; Avestro, A.-J.; Barnes, J. C.; Stoddart, J. F.; Grzybowski, B. A. Adv. Mater. 2013, 25, 331–348. doi:10.1002/adma.201201912
Return to citation in text: [1] -
Feringa, B. L. Angew. Chem., Int. Ed. 2017, 56, 11060–11078. doi:10.1002/anie.201702979
Return to citation in text: [1] -
Cheng, C.; McGonigal, P. R.; Schneebeli, S. T.; Li, H.; Vermeulen, N. A.; Ke, C.; Stoddart, J. F. Nat. Nanotechnol. 2015, 10, 547–553. doi:10.1038/nnano.2015.96
Return to citation in text: [1] -
Vella, S. J.; Tiburcio, J.; Loeb, S. J. Chem. Commun. 2007, 4752–4754. doi:10.1039/b710708k
Return to citation in text: [1] -
Hubbard, A. L.; Davidson, G. J. E.; Patel, R. H.; Wisner, J. A.; Loeb, S. J. Chem. Commun. 2004, 138–139. doi:10.1039/B312449E
Return to citation in text: [1] -
Hart, H.; Rajakumar, P. Tetrahedron 1995, 51, 1313–1336. doi:10.1016/0040-4020(94)01016-S
Return to citation in text: [1] [2] [3] -
Loeb, S. J.; Tiburcio, J.; Vella, S. J.; Wisner, J. A. Org. Biomol. Chem. 2006, 4, 667–680. doi:10.1039/b514528g
Return to citation in text: [1] [2] -
Attalla, M. I.; McAlpine, N. S.; Summers, L. A. Z. Naturforsch. 1984, 39b, 74–78. doi:10.1515/znb-1984-0113
Return to citation in text: [1] [2] -
Pan, J.; Han, X.; Sun, N.; Wu, H.; Lin, D.; Tien, P.; Zhou, H.-B.; Wu, S. RSC Adv. 2015, 5, 55100–55108. doi:10.1039/C5RA07286G
Return to citation in text: [1] [2]
| 1. | Bruns, C. J.; Stoddart, J. F. The Nature of the Mechanical Bond; John Wiley & Sons: Hoboken, New Jersey, 2017. |
| 2. | Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216 |
| 3. | Anelli, P. L.; Spencer, N.; Stoddart, J. F. J. Am. Chem. Soc. 1991, 113, 5131–5133. doi:10.1021/ja00013a096 |
| 4. | Zhu, K.; Vukotic, V. N.; Loeb, S. J. Angew. Chem., Int. Ed. 2012, 51, 2168–2172. doi:10.1002/anie.201108488 |
| 5. | Vukotic, V. N.; Zhu, K.; Baggi, G.; Loeb, S. J. Angew. Chem., Int. Ed. 2017, 56, 6136–6141. doi:10.1002/anie.201612549 |
| 17. | Hubbard, A. L.; Davidson, G. J. E.; Patel, R. H.; Wisner, J. A.; Loeb, S. J. Chem. Commun. 2004, 138–139. doi:10.1039/B312449E |
| 16. | Vella, S. J.; Tiburcio, J.; Loeb, S. J. Chem. Commun. 2007, 4752–4754. doi:10.1039/b710708k |
| 10. | Ragazzon, G.; Baroncini, M.; Silvi, S.; Venturi, M.; Credi, A. Nat. Nanotechnol. 2015, 10, 70–75. doi:10.1038/nnano.2014.260 |
| 11. | Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455 |
| 12. | Collier, C. P.; Mattersteig, G.; Wong, E. W.; Luo, Y.; Beverly, K.; Sampaio, J.; Raymo, F. M.; Stoddart, J. F.; Heath, J. R. Science 2000, 289, 1172–1175. doi:10.1126/science.289.5482.1172 |
| 13. | Fahrenbach, A. C.; Warren, S. C.; Incorvati, J. T.; Avestro, A.-J.; Barnes, J. C.; Stoddart, J. F.; Grzybowski, B. A. Adv. Mater. 2013, 25, 331–348. doi:10.1002/adma.201201912 |
| 14. | Feringa, B. L. Angew. Chem., Int. Ed. 2017, 56, 11060–11078. doi:10.1002/anie.201702979 |
| 15. | Cheng, C.; McGonigal, P. R.; Schneebeli, S. T.; Li, H.; Vermeulen, N. A.; Ke, C.; Stoddart, J. F. Nat. Nanotechnol. 2015, 10, 547–553. doi:10.1038/nnano.2015.96 |
| 6. | Bruns, C. J.; Stoddart, J. F. Acc. Chem. Res. 2014, 47, 2186–2199. doi:10.1021/ar500138u |
| 7. | Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791 |
| 8. | Thordarson, P.; Bijsterveld, E. J. A.; Rowan, A. E.; Nolte, R. J. M. Nature 2003, 424, 915–918. doi:10.1038/nature01925 |
| 9. | Xue, M.; Yang, Y.; Chi, X.; Yan, X.; Huang, F. Chem. Rev. 2015, 115, 7398–7501. doi:10.1021/cr5005869 |
| 18. | Hart, H.; Rajakumar, P. Tetrahedron 1995, 51, 1313–1336. doi:10.1016/0040-4020(94)01016-S |
| 19. | Loeb, S. J.; Tiburcio, J.; Vella, S. J.; Wisner, J. A. Org. Biomol. Chem. 2006, 4, 667–680. doi:10.1039/b514528g |
| 20. | Attalla, M. I.; McAlpine, N. S.; Summers, L. A. Z. Naturforsch. 1984, 39b, 74–78. doi:10.1515/znb-1984-0113 |
| 21. | Pan, J.; Han, X.; Sun, N.; Wu, H.; Lin, D.; Tien, P.; Zhou, H.-B.; Wu, S. RSC Adv. 2015, 5, 55100–55108. doi:10.1039/C5RA07286G |
| 21. | Pan, J.; Han, X.; Sun, N.; Wu, H.; Lin, D.; Tien, P.; Zhou, H.-B.; Wu, S. RSC Adv. 2015, 5, 55100–55108. doi:10.1039/C5RA07286G |
| 19. | Loeb, S. J.; Tiburcio, J.; Vella, S. J.; Wisner, J. A. Org. Biomol. Chem. 2006, 4, 667–680. doi:10.1039/b514528g |
| 20. | Attalla, M. I.; McAlpine, N. S.; Summers, L. A. Z. Naturforsch. 1984, 39b, 74–78. doi:10.1515/znb-1984-0113 |
| 18. | Hart, H.; Rajakumar, P. Tetrahedron 1995, 51, 1313–1336. doi:10.1016/0040-4020(94)01016-S |
| 18. | Hart, H.; Rajakumar, P. Tetrahedron 1995, 51, 1313–1336. doi:10.1016/0040-4020(94)01016-S |
© 2018 Vella and Loeb; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)