Abstract
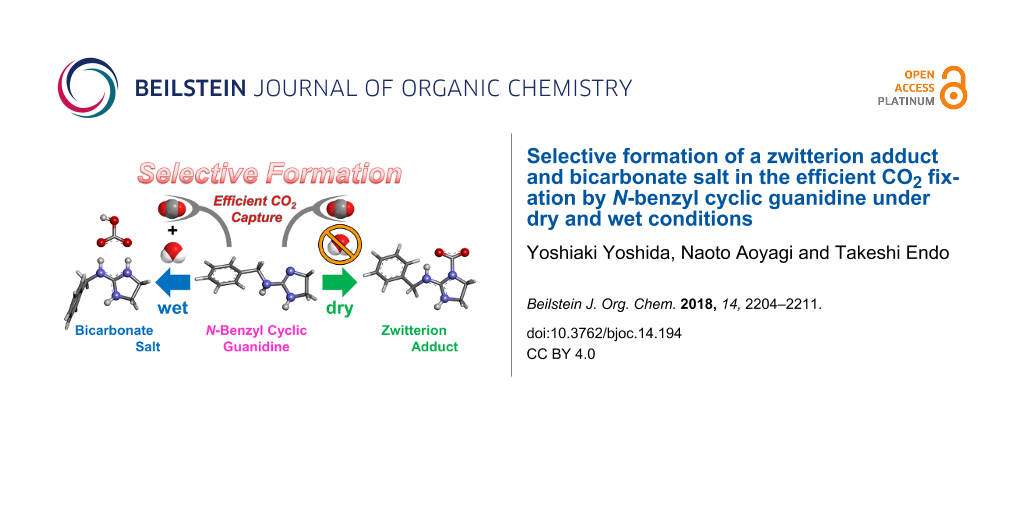
The efficient CO2 fixation by N-benzyl cyclic guanidine 1 was achieved by bubbling dry CO2 through CH3CN at 25 °C for 2 h. In addition, the zwitterion adduct 2 and bicarbonate salt 3 were selectively prepared from 1 under dry (in anhydrous CH3CN) and wet (in CH3CN containing an equimolar amount of water for 1) conditions, respectively. Both compounds 2 and 3 were isolated as white solids and their structures were characterized in detail by elemental analysis, FTIR-ATR, solid-state NMR, TGA, and DFT calculation. These analytical results obviously revealed the formation of a zwitterion adduct and bicarbonate salt from N-benzyl cyclic guanidine and CO2. Especially, the zwitterion adduct of the monocyclic guanidine derivative and CO2 was isolated and characterized for the first time.

Graphical Abstract
Introduction
Recently, various reactions with CO2 as a cheap and green carbon reagent have been developed not only in the field of organic and catalyst chemistry but also in inorganic and polymer chemistry. Especially the adsorbent and the catalyst for CO2 fixation are of interest [1-5]. In particular, the inorganic, organic–inorganic hybrid, dendrimeric catalysts have been developed for chemical fixation of CO2 such as cycloaddition of CO2 to various epoxides and synthesis of carbonate derivatives from CO2 and alcohols [6-13]. However, these kinds of catalysts often need high temperatures and pressure to completely achieve the CO2 fixation [6]. On the other hand, amidine and guanidine derivatives are well known as efficient catalysts for the cycloaddition of CO2 to epoxides at ambient temperature [7,14-18]. Previously, we reported that hydroiodides of amidine derivatives worked as significantly efficient catalysts for the cycloaddition of CO2 to epoxides under ambient temperature and pressure [19-22]. Furthermore, cyclic amidines and guanidines, such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), exhibited an excellent efficiency of CO2 capture and release [23-33]. In particular, CO2 capture–release behaviors of bicyclic and six-membered cyclic amidine derivatives have been well studied compared to that of five-membered derivatives, because the high ring strain of five-membered cyclic amidine derivatives was unfavorable for the binding between CO2 and the amidine moiety [34-38]. However, we found that five-membered cyclic guanidine was excellently efficient for CO2 capture under dry conditions because the trapped CO2 was significantly stabilized as the bicarbonate together with a slight amount of water due to the specific basicity based on the resonance structure of the guanidine moiety [39]. Furthermore, we also have demonstrated that the adsorption performance of CO2 was fairly different under dry and wet conditions [35]. Previously, Jessop et al. described that the bicarbonate salt of DBU and CO2 was obtained in the presence of water and never confirmed in the absence of water [23]. Moreover, Pérez et al. indicated that the DBU–CO2 complex was assigned as the zwitterion adduct of DBU and CO2 involving water by 13C NMR, TGA, and elemental analysis, although the bicarbonate salt was formed due to hydrolysis in the DBU–CO2 carbamic complex involving water during the crystallization process [28]. On the other hand, Villiers and Ephritikhine et al. synthesized and characterized successfully the zwitterion adduct of CO2 and TBD under strictly anhydrous conditions [31]. They also described that the obtained zwitterion adduct of TBD–CO2 was stable at room temperature in a dry atmosphere of argon for at least one month. This report is a limited example on the isolated zwitterion adduct of guanidine derivatives and CO2. As such, several researchers have demonstrated about the formation of zwitterion adduct and/or bicarbonate salt between guanidine derivatives and CO2 under dry and/or wet conditions. Previously, there were a lot of reports about the bicarbonate salt of guanidine derivatives and CO2 [23-40]. On the other hand, preparation and isolation of the zwitterion adduct have been barely reported, because the zwitterion adduct was readily transformed to the bicarbonate salt owing to ambient moisture. Therefore, it is worthwhile to synthesize and isolate the moisture stable zwitterionic adduct of guanidine derivatives and CO2, because the zwitterionic adduct contains an activated carbon dioxide without a water molecule. It is expected that the zwitterion adducts of guanidine derivatives and CO2 are not only efficient catalysts for cycloaddition of CO2 to epoxides but also a thermal latent curing agent for epoxy resin. Herein, we achieved selective formation of the zwitterionic adduct and bicarbonate salt between CO2 and N-benzyl cyclic guanidine 1. The structures of the zwitterionic adduct and bicarbonate salt were analyzed in detail and proved by elemental analysis, FTIR-ATR, solid-state NMR, TGA, and DFT calculations. In this report, the zwitterionic adduct of the monocyclic guanidine derivative and CO2 was isolated and characterized for the first time, although there were a few reports about the bicyclic guanidine derivatives such as TBD.
Results and Discussion
CO2 fixation behavior of N-benzyl cyclic guanidine depending on dry and wet conditions
First, CO2 fixation by N-benzyl cyclic guanidine 1 was carried out by bubbling dry CO2 through anhydrous CH3CN unter dry conditions (Scheme 1a) and through CH3CN containing an equimolar amount of water for 1 as wet condition (Scheme 1b) at 25 °C for 2 h. The white solid was immediately precipitated in the homogeneous solution of 1 during CO2 bubbling under both conditions. The efficient absorption of CO2 by 1 suggested that the trapped CO2 was stabilized by the active hydrogen on the nitrogen atom. This hydrogen was fairly activated due to the resonance effect of the guanidine moiety [16,39]. The compositional formula of the obtained white solids was confirmed by elemental analysis. This result predicted that the zwitterionic adduct 2 and the bicarbonate salt 3 were prepared selectively under dry and wet conditions, respectively (Figure 1). Interestingly, 3 was barely obtained by the addition of an equivalent water to the precipitation of 2 in CH3CN, and then the only part of 2 was transformed to 3 after stirring in CH3CN containing an excess mole of water, although 2 and 3 were insoluble in organic solvents. This result means that 2 is hydrophobic enough to be isolated as an individual compound. On the other hand, 3 was also prepared from 2 dissolved completely in water, because 1H NMR spectra of 2 and 3 showed the same signals in D2O (Supporting Information File 1, section 2-1) and the carbonyl peaks due to the bicarbonate moiety were observed at 160.8 ppm in D2O by 13C NMR spectra of 2 and 3 (Figure 2).
![[1860-5397-14-194-i1]](/bjoc/content/inline/1860-5397-14-194-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fixation of CO2 (200 mL/min) by 1 under (a) dry and (b) wet conditions.
Scheme 1: Fixation of CO2 (200 mL/min) by 1 under (a) dry and (b) wet conditions.
![[1860-5397-14-194-1]](/bjoc/content/figures/1860-5397-14-194-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Zwitterion adduct 2 and bicarbonate salt 3 confirmed by elemental analysis.
Figure 1: Zwitterion adduct 2 and bicarbonate salt 3 confirmed by elemental analysis.
![[1860-5397-14-194-2]](/bjoc/content/figures/1860-5397-14-194-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 13C NMR spectra of (a) 1 observed in DMSO-d6, (b) 3' prepared with 2 and D2O observed in D2O, and (c) 3 prepared with 1 under wet conditions observed in D2O.
Figure 2: 13C NMR spectra of (a) 1 observed in DMSO-d6, (b) 3' prepared with 2 and D2O observed in D2O, and (...
Characterization of zwitterion adduct 2 and bicarbonate salt 3 by FTIR-ATR and solid-state 13C-CPMAS NMR
IR spectra of 2 and 3 were measured by FTIR-ATR methods (Figure 3). The two peaks were clearly observed at 1573 cm−1 and 1706 cm−1 due to carbonyl and imine moieties, respectively, in the spectrum of 2. On the other hand, the peak at 1593 cm−1 was assigned to the carbonyl group of the bicarbonate moiety, and also the peaks due to C=N and N–H of iminium group in guanidinium moiety were observed at 1677 cm−1 and 1621 cm−1, respectively, in the spectrum of 3.
![[1860-5397-14-194-3]](/bjoc/content/figures/1860-5397-14-194-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: FTIR-ATR spectra of zwitterion adduct 2 and bicarbonate salt 3 expanded at the range of 1500–1800 cm−1.
Figure 3: FTIR-ATR spectra of zwitterion adduct 2 and bicarbonate salt 3 expanded at the range of 1500–1800 cm...
Moreover, the structures of 2 and 3 were also revealed by 13C-CPMAS NMR in solid-state (Figure 4). The carbon peaks due to the trapped CO2 and guanidinium moieties were observed at 135.4 ppm and 157.2 ppm, respectively, in the spectrum of 2, which indicated the zwitterionic structure between CO2 and 1. On the other hand, the carbon peaks due to the bicarbonate and guanidinium moieties were observed at 161.3 ppm and 163.1 ppm, respectively, in the spectrum of 3. This chemical shift of 2 suggested that the electron density on the carbonyl carbon of 2 increased by the donation of an electron from the imine nitrogen, and also the electron at the iminium carbon was localized due to the imine structure stabilized by the direct binding with CO2. Therefore, both the peaks of carbonyl and iminium carbons in 2 shifted to higher magnetic field compared to that of 3. On the other hand, both the peaks of bicarbonate and guanidinium carbons in 3 were observed at a low magnetic field, because the electrons on bicarbonate and guanidinium carbons were delocalized on oxygen and nitrogen atoms by their resonance effect, respectively. Previously, some researchers have reported similar assignments for zwitterion adducts of amidines and guanidines [29-31]. The IR and NMR spectra indicated that 2 and 3 were selectively prepared under dry and wet conditions, respectively, and then 2 and 3 were obtained as fairly stable compounds under ambient temperature and moisture.
![[1860-5397-14-194-4]](/bjoc/content/figures/1860-5397-14-194-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 13C-CPMAS NMR spectra of zwitterion adduct 2 and bicarbonate salt 3 expanded at the range of 30–170 ppm.
Figure 4: 13C-CPMAS NMR spectra of zwitterion adduct 2 and bicarbonate salt 3 expanded at the range of 30–170...
Computational evaluation for the CO2 fixation behavior of N-benzyl cyclic guanidine
The geometrical structures of 2 and 3 were computed with DFT calculation (Figure 5). In the calculated geometry for 2, the bond length between CO2 and imine nitrogen was 1.548 Å and the bent angle of O–C–O bond was 135.76°. These bond length and angle were fairly similar to that of the zwitterion adduct between CO2 and TBD calculated by Wei and Sun et al. [40]. Moreover, the distance between the trapped CO2 and the active hydrogen at the nitrogen atom was close to each other (1.686 Å), which also agreed with that of TBD–CO2 adduct calculated by Villiers and Ephritikhine et al. [31]. On the other hand, the calculated geometry for 3 showed that the distance between the two protons of the imidazolinium moiety and two oxygen atoms of the bicarbonate moiety were fairly close to each other (1.562 Å and 1.578 Å), indicating that the two oxygen atoms on the bicarbonate are stabilized by the active hydrogen on the imidazolinium moiety. Furthermore, due to the guanidine moiety, three nitrogen atoms of 2 and 3 were in a more planar position than that of 1, which was the twisted conformation between the benzylamino group and the cyclic amidine moiety (Supporting Information File 1, section 1-1). Therefore, these results strongly supported the geometrical structures and electronic state of 2 and 3 predicted by 13C NMR and IR spectra.
![[1860-5397-14-194-5]](/bjoc/content/figures/1860-5397-14-194-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The optimized geometries of zwitterion adduct 2 and bicarbonate salt 3 estimated by DFT calculation (B3LYP/6-31G*).
Figure 5: The optimized geometries of zwitterion adduct 2 and bicarbonate salt 3 estimated by DFT calculation...
Thermal decomposition behaviors of zwitterion adduct 2 and bicarbonate salt 3
Thermal decomposition behaviors of 2 and 3 were observed by TGA measurement. TGA traces and weight loss values of 2 and 3 are shown in Figure 6. Moreover, their proposed decomposition paths and the theoretical values of weight loss are shown in Scheme 2. The weight loss of 2 exhibited a two-step behavior before complete thermal decomposition (Figure 6a). Both the first and second weight loss proceeded slowly until 102 °C (i) and 173 °C (ii), respectively, and then the thermal decomposition of 2 has occurred completely around 270 °C (iii). This three-step behavior of weight loss suggested the dimer form of 2, which was constructed by the weak interaction between the anionic oxygen on the trapped CO2 and the active hydrogen on the nitrogen atom. Namely, one molecule of CO2 was released from the dimer of 2 in the first step, and then the intermediate complex was prepared from 1 and 2 due to the coordination between two anionic oxygens on the trapped CO2 and two active hydrogens on the guanidine moiety. Then, another CO2 molecule was released from the intermediate complex (2+1 dimer) and the dimer was decomposed into two molecules of 1 in the second step. The observed weight loss due to the released CO2 in each step agreed with the theoretical one (obs. (i) 10.3%, (ii) 10.6%, and (iii) 78.4%, theor. (i) 10.1%, (ii) 10.1%, and (iii) 79.8%). On the other hand, the weight loss of 3 also exhibited a three-step behavior as well as 2, although the TGA trace pattern was perfectly different from that of 2 (Figure 6b). The first weight loss occurred rapidly around 90 °C (until 96 °C) (i), and then the second weight loss exhibited rapid and slow weight loss behaviors around 127 °C and until 162 °C (ii), respectively. Finally, the thermal decomposition of 3 has occurred completely around 260 °C (iii). This three-steps behavior of weight loss suggested that the dimer of 3 was constructed by the two hydrogen bonds between two oxygen atoms in two HCO3− anions. Recently, we demonstrated that the bicarbonate salt of the cyclic guanidine derivative was decomposed at three-steps [39]. Similarly, the first weight loss until 96 °C was assigned to the elimination of one pair of CO2 and H2O due to the bicarbonate moiety in the dimer of 3. Accordingly, the intermediate complex of 3 and 1 was constructed by the two hydrogen bonds between OH−N and O−HN in the bicarbonate salt 3 and the free guanidine 1, and then another one pair of CO2 and H2O was almost released from the intermediate complex (3+1 dimer) around 127 °C in the second step. After the rapid elimination of CO2 and H2O around 127 °C, the remained bicarbonate moiety was also released slowly from 3+1 dimer until 162 °C, because the bicarbonate moiety was probably stabilized by a large amount of the free guanidine. The observed weight loss from the dimer of 3 also agreed with the theoretical one as well as 2 (obs. (i) 12.2%, (ii) 13.9%, and (iii) 72.5%, theor. (i) 13.1%, (ii) 13.1%, and (iii) 73.8%). Therefore, this thermal decomposition behavior indicated that the dimers of zwitterion adduct and bicarbonate salt were constructed in solid-state and their structure was fairly stabilized because of the multi hydrogen bond in the intermolecular.
![[1860-5397-14-194-6]](/bjoc/content/figures/1860-5397-14-194-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: TGA trace of (a) zwitterion adduct 2 and (b) bicarbonate salt 3 observed under N2 flow (200 mL/min) at heating rate of 5 °C/min.
Figure 6: TGA trace of (a) zwitterion adduct 2 and (b) bicarbonate salt 3 observed under N2 flow (200 mL/min)...
![[1860-5397-14-194-i2]](/bjoc/content/inline/1860-5397-14-194-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposal decomposition paths and theoretical weight loss values of (a) zwitterion adduct 2 and (b) bicarbonate salt 3.
Scheme 2: Proposal decomposition paths and theoretical weight loss values of (a) zwitterion adduct 2 and (b) ...
Conclusion
The efficient CO2 fixation by N-benzyl cyclic guanidine 1 was achieved by bubbling dry CO2 in CH3CN at 25 °C for 2 h. In addition, the zwitterion adduct 2 and bicarbonate salt 3 were selectively prepared from 1 under dry (in anhydrous CH3CN) and wet (in CH3CN containing an equimolar amount of water for 1) conditions, respectively. Both of 2 and 3 were isolated as white solids and their structures were characterized in detail by elemental analysis, FTIR-ATR, solid-state NMR, TGA, and DFT calculation. Especially, the results of the thermal analysis revealed that the obtained zwitterion adduct and bicarbonate salt stabilized the dimer complexes based on multiple intermolecular hydrogen bonds. This also indicated that the zwitterion adduct, bicarbonate salt, and their dimers were a fairly stable structure at room temperature and ambient moisture.
Experimental
General
Materials and instruments
All starting materials and dehydrated solvents were purchased from Wako Pure Chemical Industries (Osaka, Japan) and Tokyo Chemical Industry (Tokyo, Japan). CO2 (>99.999%, H2O < 5 ppm) was obtained from Yamagata Sanso (Yamagata, Japan). The NMR spectra were obtained using a JEOL ECS-400 spectrometer operating at 400 MHz for 1H and 100 MHz for 13C. The elemental analyses were performed by a Perkin Elmer 2400II CHNS/O Analyzer. Mass spectroscopy was performed on a Shimadzu GCMS-QP2010SE in electron ionization (EI) mode. FTIR spectra were recorded on a Thermo Scientific Nicolet iS10 spectrometer equipped with a Smart iTR diamond ATR sampling accessory in the range of 4000–650 cm−1. Solid-state NMR, 13C cross-polarization (CP)/magic angle spinning (MAS, 99.5 MHz) measurements were performed on a JEOL JNM-ECX 400 spectrometer, at a spinning speed of 10 kHz. Thermogravimetric analysis (TGA) was performed on a Seiko Instrument TG-DTA 6200 using an aluminum pan in the temperature range of 30–350 °C at a heating rate of 5 °C/min under a nitrogen atmosphere (flow rate 200 mL/min). Geometry optimized energy for 1, 2 and 3 was estimated by using DFT calculation with B3LYP/6-31G* method (Wavefunction, Inc., Spartan’06 Windows version 1.1.0) [41].
Synthesis of N-benzyl cyclic guanidine
2-Benzylamino-4,5-dihydro-1H-imidazole (N-benzyl cyclic guanidine, 1) was synthesized according to the literature [42]. mp 79.9–83.1 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C) δ (ppm) 7.20–7.33 (m, 7H), 4.26 (s, 2H), 3.37 (s, 4H); 13C NMR (100 MHz, DMSO-d6, 25 °C) δ (ppm) 161.3, 140.3, 128.0, 127.1, 126.5, 46.5, 46.0; EIMS (m/z): 175 (M+).
Procedure for CO2 fixation under dry and wet conditions [39]
Dry CO2 gas (>99.999%, H2O <5 ppm) was bubbled into a solution of 1 (4 mmol) in anhydrous CH3CN (10 mL) at a flow rate of 200 mL/min at 25 ºC. After 2 h under bubbling CO2, the resulting white precipitate was filtered off, washed with anhydrous Et2O (10 mL × 3) and dried in a stream of CO2 (at a flow rate of 200 mL/min at 25 °C for 4 h) to give zwitterion adduct 2 as a white powder.
Zwitterion adduct 2: Yield 798 mg, 91%; mp 92.1–109.4 °C; IR (ATR, cm−1) 1706 (C=N), 1573 (C=O); 13C-CPMAS NMR (99.5 MHz, 25 °C) δ (ppm) 157.2 (C=N), 137.3 (aromatic), 135.4 (CO2), 129.5 (aromatic), 48.6 and 47.1 (NCH2CH2NH), 42.6 (CH2Ph); anal. calcd for C11H13N3O2: C, 60.26; H, 5.98; N, 19.17; found: C, 60.10; H, 5.98; N, 19.20.
CO2 fixation of 1 under wet conditions
Dry CO2 gas (>99.999%, H2O <5 ppm) was bubbled into a solution of 1 (4 mmol) in CH3CN (10 mL) containing water (0.1 mL) at a flow rate of 200 mL/min at 25 ºC. After 2 h under bubbling CO2, the resulting white precipitate was filtered off, washed with anhydrous Et2O (10 mL × 3) and dried in a stream of CO2 (at a flow rate of 200 mL/min at 25 ºC for 4 h) to give bicarbonate salt 3 as a white powder.
Bicarbonate salt 3: Yield 930 mg, 98%; mp 89.7–109.2 °C; IR (ATR, cm−1) 1677 (C=N), 1621 (C=N−H), 1593 (C=O); 13C-CPMAS NMR (99.5 MHz, 25 °C) δ (ppm) 163.1 (C=N), 161.3 (HCO3), 139.8 and 127.7 (aromatic), 45.7 (NCH2CH2NH), 43.5 (CH2Ph); anal. calcd for C11H15N3O3: C, 55.69; H, 6.37; N, 17.71; found: C, 55.48; H, 6.29; N, 17.84.
Supporting Information
| Supporting Information File 1: DFT computational results, FTIR-ATR, and NMR spectra of 1, 2 and 3. | ||
| Format: PDF | Size: 732.0 KB | Download |
References
-
Kango, S.; Kalia, S.; Celli, A.; Njuguna, J.; Habibi, Y.; Kumar, R. Prog. Polym. Sci. 2013, 38, 1232–1261. doi:10.1016/j.progpolymsci.2013.02.003
Return to citation in text: [1] -
Ganesh, I. Renewable Sustainable Energy Rev. 2014, 31, 221–257. doi:10.1016/j.rser.2013.11.045
Return to citation in text: [1] -
Razali, N. A. M.; Lee, K. T.; Bhatia, S.; Mohamed, A. R. Renewable Sustainable Energy Rev. 2012, 16, 4951–4964. doi:10.1016/j.rser.2012.04.012
Return to citation in text: [1] -
Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. Chem. Soc. Rev. 2014, 43, 631–675. doi:10.1039/C3CS60323G
Return to citation in text: [1] -
Wang, W.; Wang, S.; Ma, X.; Gong, J. Chem. Soc. Rev. 2011, 40, 3703–3727. doi:10.1039/c1cs15008a
Return to citation in text: [1] -
Mikkelsen, M.; Jørgensen, M.; Krebs, F. C. Energy Environ. Sci. 2010, 3, 43–81. doi:10.1039/B912904A
Return to citation in text: [1] [2] -
Shaikh, R. R.; Pornpraprom, S.; D’Elia, V. ACS Catal. 2018, 8, 419–450. doi:10.1021/acscatal.7b03580
Return to citation in text: [1] [2] -
Miralda, C. M.; Macias, E. E.; Zhu, M.; Ratnasamy, P.; Carreon, M. A. ACS Catal. 2012, 2, 180–183. doi:10.1021/cs200638h
Return to citation in text: [1] -
Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J. S.; Zou, R.; Zhao, Y. J. Am. Chem. Soc. 2016, 138, 2142–2145. doi:10.1021/jacs.5b13335
Return to citation in text: [1] -
Zou, C.; Zhang, Z.; Xu, X.; Gong, Q.; Li, J.; Wu, C.-D. J. Am. Chem. Soc. 2012, 134, 87–90. doi:10.1021/ja209196t
Return to citation in text: [1] -
Ema, T.; Miyazaki, Y.; Shimonishi, J.; Maeda, C.; Hasegawa, J. J. Am. Chem. Soc. 2014, 136, 15270–15279. doi:10.1021/ja507665a
Return to citation in text: [1] -
Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G. A.; Prakash, G. K. S. J. Am. Chem. Soc. 2016, 138, 778–781. doi:10.1021/jacs.5b12354
Return to citation in text: [1] -
Lin, J.; Pan, Z.; Wang, X. ACS Sustainable Chem. Eng. 2014, 2, 353–358. doi:10.1021/sc4004295
Return to citation in text: [1] -
Quek, J. Y.; Davis, T. P.; Lowe, A. B. Chem. Soc. Rev. 2013, 42, 7326–7334. doi:10.1039/c3cs60065c
Return to citation in text: [1] -
Castagnolo, D.; Schenone, S.; Botta, M. Chem. Rev. 2011, 111, 5247–5300. doi:10.1021/cr100423x
Return to citation in text: [1] -
Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/c2cs15288f
Return to citation in text: [1] [2] -
Lin, S.; Theato, P. Macromol. Rapid Commun. 2013, 34, 1118–1133. doi:10.1002/marc.201300288
Return to citation in text: [1] -
Liu, H.; Lin, S.; Feng, Y.; Theato, P. Polym. Chem. 2017, 8, 12–23. doi:10.1039/C6PY01101B
Return to citation in text: [1] -
Barkakaty, B.; Morino, K.; Sudo, A.; Endo, T. Green Chem. 2010, 12, 42–44. doi:10.1039/B916235F
Return to citation in text: [1] -
Aoyagi, N.; Furusho, Y.; Endo, T. Chem. Lett. 2012, 41, 240–241. doi:10.1246/cl.2012.240
Return to citation in text: [1] -
Aoyagi, N.; Furusho, Y.; Endo, T. Tetrahedron Lett. 2013, 54, 7031–7034. doi:10.1016/j.tetlet.2013.10.068
Return to citation in text: [1] -
Aoyagi, N.; Furusho, Y.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1230–1242. doi:10.1002/pola.26492
Return to citation in text: [1] -
Heldebrant, D. J.; Jessop, P. G.; Thomas, C. A.; Eckert, C. A.; Liotta, C. L. J. Org. Chem. 2005, 70, 5335–5338. doi:10.1021/jo0503759
Return to citation in text: [1] [2] [3] -
Jessop, P. G.; Heldebrant, D. J.; Li, X.; Eckert, C. A.; Liotta, C. L. Nature 2005, 436, 1102. doi:10.1038/4361102a
Return to citation in text: [1] [2] -
Liu, Y.; Jessop, P. G.; Cunningham, M.; Eckert, C. A.; Liotta, C. L. Science 2006, 313, 958–960. doi:10.1126/science.1128142
Return to citation in text: [1] [2] -
Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Environ. Sci. 2008, 1, 487–493. doi:10.1039/B809533G
Return to citation in text: [1] [2] -
Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Procedia 2009, 1, 1187–1195. doi:10.1016/j.egypro.2009.01.156
Return to citation in text: [1] [2] -
Pérez, E. R.; Santos, R. H. A.; Gambardella, M. T. P.; de Macedo, L. G. M.; Rodrigues-Filho, U. P.; Launay, J.-C.; Franco, D. W. J. Org. Chem. 2004, 69, 8005–8011. doi:10.1021/jo049243q
Return to citation in text: [1] [2] [3] -
Pereira, F. S.; deAzevedo, E. R.; da Silva, E. F.; Bonagamba, T. J.; da Silva Agostíni, D. L.; Magalhães, A.; Job, A. E.; Pérez González, E. R. Tetrahedron 2008, 64, 10097–10106. doi:10.1016/j.tet.2008.08.008
Return to citation in text: [1] [2] [3] -
Pereira, F. S.; da Silva Agostini, D. L.; do Espírito Santo, R. D.; deAzevedo, E. R.; Bonagamba, T. J.; Job, A. E.; Pérez González, E. R. Green Chem. 2011, 13, 2146–2153. doi:10.1039/c1gc15457e
Return to citation in text: [1] [2] [3] -
Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035
Return to citation in text: [1] [2] [3] [4] [5] -
Heldebrant, D. J.; Koech, P. K.; Rainbolt, J.; Zheng, F.; Smurthwaite, T.; Freeman, C. J.; Oss, M.; Leito, I. Chem. Eng. J. 2011, 171, 794–800. doi:10.1016/j.cej.2011.02.012
Return to citation in text: [1] [2] -
Koech, P. K.; Zhang, J.; Kutnyakov, I. V.; Cosimbescu, L.; Lee, S.-J.; Bowden, M. E.; Smurthwaite, T. D.; Heldebrant, D. J. RSC Adv. 2013, 3, 566–572. doi:10.1039/C2RA22801G
Return to citation in text: [1] [2] -
Aoyagi, N.; Furusho, Y.; Sei, Y.; Endo, T. Tetrahedron 2013, 69, 5476–5480. doi:10.1016/j.tet.2013.04.110
Return to citation in text: [1] [2] -
Aoyagi, N.; Endo, T. Tetrahedron 2017, 73, 1529–1533. doi:10.1016/j.tet.2017.01.012
Return to citation in text: [1] [2] [3] -
Endo, T.; Nagai, D.; Monma, T.; Yamaguchi, H.; Ochiai, B. Macromolecules 2004, 37, 2007–2009. doi:10.1021/ma0305479
Return to citation in text: [1] [2] -
Ochiai, B.; Yokota, K.; Fujii, A.; Nagai, D.; Endo, T. Macromolecules 2008, 41, 1229–1236. doi:10.1021/ma702189a
Return to citation in text: [1] [2] -
Sakuragi, M.; Aoyagi, N.; Furusho, Y.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 2025–2031. doi:10.1002/pola.27210
Return to citation in text: [1] [2] -
Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E
Return to citation in text: [1] [2] [3] [4] [5] -
Ma, J.; Zhang, X.; Zhao, N.; Al-Arifi, A. S. N.; Aouak, T.; Al-Othman, Z. A.; Xiao, F.; Wei, W.; Sun, Y. J. Mol. Catal. A: Chem. 2010, 315, 76–81. doi:10.1016/j.molcata.2009.09.003
Return to citation in text: [1] [2] -
Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O’Neill, D. P.; DiStasio, R. A., Jr.; Lochan, R. C.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Voorhis, T. V.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C.-P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock, H. L., III; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. I.; Warshel, A.; Hehre, W. J.; Schaefer, H. F., III; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. doi:10.1039/B517914A
Return to citation in text: [1] -
Aoyagi, N.; Endo, T. Synth. Commun. 2017, 47, 442–448. doi:10.1080/00397911.2016.1269927
Return to citation in text: [1]
| 41. | Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O’Neill, D. P.; DiStasio, R. A., Jr.; Lochan, R. C.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Voorhis, T. V.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C.-P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock, H. L., III; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. I.; Warshel, A.; Hehre, W. J.; Schaefer, H. F., III; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. doi:10.1039/B517914A |
| 31. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 39. | Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E |
| 1. | Kango, S.; Kalia, S.; Celli, A.; Njuguna, J.; Habibi, Y.; Kumar, R. Prog. Polym. Sci. 2013, 38, 1232–1261. doi:10.1016/j.progpolymsci.2013.02.003 |
| 2. | Ganesh, I. Renewable Sustainable Energy Rev. 2014, 31, 221–257. doi:10.1016/j.rser.2013.11.045 |
| 3. | Razali, N. A. M.; Lee, K. T.; Bhatia, S.; Mohamed, A. R. Renewable Sustainable Energy Rev. 2012, 16, 4951–4964. doi:10.1016/j.rser.2012.04.012 |
| 4. | Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. Chem. Soc. Rev. 2014, 43, 631–675. doi:10.1039/C3CS60323G |
| 5. | Wang, W.; Wang, S.; Ma, X.; Gong, J. Chem. Soc. Rev. 2011, 40, 3703–3727. doi:10.1039/c1cs15008a |
| 19. | Barkakaty, B.; Morino, K.; Sudo, A.; Endo, T. Green Chem. 2010, 12, 42–44. doi:10.1039/B916235F |
| 20. | Aoyagi, N.; Furusho, Y.; Endo, T. Chem. Lett. 2012, 41, 240–241. doi:10.1246/cl.2012.240 |
| 21. | Aoyagi, N.; Furusho, Y.; Endo, T. Tetrahedron Lett. 2013, 54, 7031–7034. doi:10.1016/j.tetlet.2013.10.068 |
| 22. | Aoyagi, N.; Furusho, Y.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1230–1242. doi:10.1002/pola.26492 |
| 29. | Pereira, F. S.; deAzevedo, E. R.; da Silva, E. F.; Bonagamba, T. J.; da Silva Agostíni, D. L.; Magalhães, A.; Job, A. E.; Pérez González, E. R. Tetrahedron 2008, 64, 10097–10106. doi:10.1016/j.tet.2008.08.008 |
| 30. | Pereira, F. S.; da Silva Agostini, D. L.; do Espírito Santo, R. D.; deAzevedo, E. R.; Bonagamba, T. J.; Job, A. E.; Pérez González, E. R. Green Chem. 2011, 13, 2146–2153. doi:10.1039/c1gc15457e |
| 31. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 7. | Shaikh, R. R.; Pornpraprom, S.; D’Elia, V. ACS Catal. 2018, 8, 419–450. doi:10.1021/acscatal.7b03580 |
| 14. | Quek, J. Y.; Davis, T. P.; Lowe, A. B. Chem. Soc. Rev. 2013, 42, 7326–7334. doi:10.1039/c3cs60065c |
| 15. | Castagnolo, D.; Schenone, S.; Botta, M. Chem. Rev. 2011, 111, 5247–5300. doi:10.1021/cr100423x |
| 16. | Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/c2cs15288f |
| 17. | Lin, S.; Theato, P. Macromol. Rapid Commun. 2013, 34, 1118–1133. doi:10.1002/marc.201300288 |
| 18. | Liu, H.; Lin, S.; Feng, Y.; Theato, P. Polym. Chem. 2017, 8, 12–23. doi:10.1039/C6PY01101B |
| 40. | Ma, J.; Zhang, X.; Zhao, N.; Al-Arifi, A. S. N.; Aouak, T.; Al-Othman, Z. A.; Xiao, F.; Wei, W.; Sun, Y. J. Mol. Catal. A: Chem. 2010, 315, 76–81. doi:10.1016/j.molcata.2009.09.003 |
| 6. | Mikkelsen, M.; Jørgensen, M.; Krebs, F. C. Energy Environ. Sci. 2010, 3, 43–81. doi:10.1039/B912904A |
| 23. | Heldebrant, D. J.; Jessop, P. G.; Thomas, C. A.; Eckert, C. A.; Liotta, C. L. J. Org. Chem. 2005, 70, 5335–5338. doi:10.1021/jo0503759 |
| 24. | Jessop, P. G.; Heldebrant, D. J.; Li, X.; Eckert, C. A.; Liotta, C. L. Nature 2005, 436, 1102. doi:10.1038/4361102a |
| 25. | Liu, Y.; Jessop, P. G.; Cunningham, M.; Eckert, C. A.; Liotta, C. L. Science 2006, 313, 958–960. doi:10.1126/science.1128142 |
| 26. | Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Environ. Sci. 2008, 1, 487–493. doi:10.1039/B809533G |
| 27. | Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Procedia 2009, 1, 1187–1195. doi:10.1016/j.egypro.2009.01.156 |
| 28. | Pérez, E. R.; Santos, R. H. A.; Gambardella, M. T. P.; de Macedo, L. G. M.; Rodrigues-Filho, U. P.; Launay, J.-C.; Franco, D. W. J. Org. Chem. 2004, 69, 8005–8011. doi:10.1021/jo049243q |
| 29. | Pereira, F. S.; deAzevedo, E. R.; da Silva, E. F.; Bonagamba, T. J.; da Silva Agostíni, D. L.; Magalhães, A.; Job, A. E.; Pérez González, E. R. Tetrahedron 2008, 64, 10097–10106. doi:10.1016/j.tet.2008.08.008 |
| 30. | Pereira, F. S.; da Silva Agostini, D. L.; do Espírito Santo, R. D.; deAzevedo, E. R.; Bonagamba, T. J.; Job, A. E.; Pérez González, E. R. Green Chem. 2011, 13, 2146–2153. doi:10.1039/c1gc15457e |
| 31. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 32. | Heldebrant, D. J.; Koech, P. K.; Rainbolt, J.; Zheng, F.; Smurthwaite, T.; Freeman, C. J.; Oss, M.; Leito, I. Chem. Eng. J. 2011, 171, 794–800. doi:10.1016/j.cej.2011.02.012 |
| 33. | Koech, P. K.; Zhang, J.; Kutnyakov, I. V.; Cosimbescu, L.; Lee, S.-J.; Bowden, M. E.; Smurthwaite, T. D.; Heldebrant, D. J. RSC Adv. 2013, 3, 566–572. doi:10.1039/C2RA22801G |
| 34. | Aoyagi, N.; Furusho, Y.; Sei, Y.; Endo, T. Tetrahedron 2013, 69, 5476–5480. doi:10.1016/j.tet.2013.04.110 |
| 35. | Aoyagi, N.; Endo, T. Tetrahedron 2017, 73, 1529–1533. doi:10.1016/j.tet.2017.01.012 |
| 36. | Endo, T.; Nagai, D.; Monma, T.; Yamaguchi, H.; Ochiai, B. Macromolecules 2004, 37, 2007–2009. doi:10.1021/ma0305479 |
| 37. | Ochiai, B.; Yokota, K.; Fujii, A.; Nagai, D.; Endo, T. Macromolecules 2008, 41, 1229–1236. doi:10.1021/ma702189a |
| 38. | Sakuragi, M.; Aoyagi, N.; Furusho, Y.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 2025–2031. doi:10.1002/pola.27210 |
| 39. | Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E |
| 40. | Ma, J.; Zhang, X.; Zhao, N.; Al-Arifi, A. S. N.; Aouak, T.; Al-Othman, Z. A.; Xiao, F.; Wei, W.; Sun, Y. J. Mol. Catal. A: Chem. 2010, 315, 76–81. doi:10.1016/j.molcata.2009.09.003 |
| 6. | Mikkelsen, M.; Jørgensen, M.; Krebs, F. C. Energy Environ. Sci. 2010, 3, 43–81. doi:10.1039/B912904A |
| 7. | Shaikh, R. R.; Pornpraprom, S.; D’Elia, V. ACS Catal. 2018, 8, 419–450. doi:10.1021/acscatal.7b03580 |
| 8. | Miralda, C. M.; Macias, E. E.; Zhu, M.; Ratnasamy, P.; Carreon, M. A. ACS Catal. 2012, 2, 180–183. doi:10.1021/cs200638h |
| 9. | Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J. S.; Zou, R.; Zhao, Y. J. Am. Chem. Soc. 2016, 138, 2142–2145. doi:10.1021/jacs.5b13335 |
| 10. | Zou, C.; Zhang, Z.; Xu, X.; Gong, Q.; Li, J.; Wu, C.-D. J. Am. Chem. Soc. 2012, 134, 87–90. doi:10.1021/ja209196t |
| 11. | Ema, T.; Miyazaki, Y.; Shimonishi, J.; Maeda, C.; Hasegawa, J. J. Am. Chem. Soc. 2014, 136, 15270–15279. doi:10.1021/ja507665a |
| 12. | Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G. A.; Prakash, G. K. S. J. Am. Chem. Soc. 2016, 138, 778–781. doi:10.1021/jacs.5b12354 |
| 13. | Lin, J.; Pan, Z.; Wang, X. ACS Sustainable Chem. Eng. 2014, 2, 353–358. doi:10.1021/sc4004295 |
| 16. | Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/c2cs15288f |
| 39. | Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E |
| 35. | Aoyagi, N.; Endo, T. Tetrahedron 2017, 73, 1529–1533. doi:10.1016/j.tet.2017.01.012 |
| 28. | Pérez, E. R.; Santos, R. H. A.; Gambardella, M. T. P.; de Macedo, L. G. M.; Rodrigues-Filho, U. P.; Launay, J.-C.; Franco, D. W. J. Org. Chem. 2004, 69, 8005–8011. doi:10.1021/jo049243q |
| 39. | Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E |
| 31. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 34. | Aoyagi, N.; Furusho, Y.; Sei, Y.; Endo, T. Tetrahedron 2013, 69, 5476–5480. doi:10.1016/j.tet.2013.04.110 |
| 35. | Aoyagi, N.; Endo, T. Tetrahedron 2017, 73, 1529–1533. doi:10.1016/j.tet.2017.01.012 |
| 36. | Endo, T.; Nagai, D.; Monma, T.; Yamaguchi, H.; Ochiai, B. Macromolecules 2004, 37, 2007–2009. doi:10.1021/ma0305479 |
| 37. | Ochiai, B.; Yokota, K.; Fujii, A.; Nagai, D.; Endo, T. Macromolecules 2008, 41, 1229–1236. doi:10.1021/ma702189a |
| 38. | Sakuragi, M.; Aoyagi, N.; Furusho, Y.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 2025–2031. doi:10.1002/pola.27210 |
| 42. | Aoyagi, N.; Endo, T. Synth. Commun. 2017, 47, 442–448. doi:10.1080/00397911.2016.1269927 |
| 23. | Heldebrant, D. J.; Jessop, P. G.; Thomas, C. A.; Eckert, C. A.; Liotta, C. L. J. Org. Chem. 2005, 70, 5335–5338. doi:10.1021/jo0503759 |
| 24. | Jessop, P. G.; Heldebrant, D. J.; Li, X.; Eckert, C. A.; Liotta, C. L. Nature 2005, 436, 1102. doi:10.1038/4361102a |
| 25. | Liu, Y.; Jessop, P. G.; Cunningham, M.; Eckert, C. A.; Liotta, C. L. Science 2006, 313, 958–960. doi:10.1126/science.1128142 |
| 26. | Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Environ. Sci. 2008, 1, 487–493. doi:10.1039/B809533G |
| 27. | Heldebrant, D. J.; Yonker, C. R.; Jessop, P. G.; Phan, L. Energy Procedia 2009, 1, 1187–1195. doi:10.1016/j.egypro.2009.01.156 |
| 28. | Pérez, E. R.; Santos, R. H. A.; Gambardella, M. T. P.; de Macedo, L. G. M.; Rodrigues-Filho, U. P.; Launay, J.-C.; Franco, D. W. J. Org. Chem. 2004, 69, 8005–8011. doi:10.1021/jo049243q |
| 29. | Pereira, F. S.; deAzevedo, E. R.; da Silva, E. F.; Bonagamba, T. J.; da Silva Agostíni, D. L.; Magalhães, A.; Job, A. E.; Pérez González, E. R. Tetrahedron 2008, 64, 10097–10106. doi:10.1016/j.tet.2008.08.008 |
| 30. | Pereira, F. S.; da Silva Agostini, D. L.; do Espírito Santo, R. D.; deAzevedo, E. R.; Bonagamba, T. J.; Job, A. E.; Pérez González, E. R. Green Chem. 2011, 13, 2146–2153. doi:10.1039/c1gc15457e |
| 31. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 32. | Heldebrant, D. J.; Koech, P. K.; Rainbolt, J.; Zheng, F.; Smurthwaite, T.; Freeman, C. J.; Oss, M.; Leito, I. Chem. Eng. J. 2011, 171, 794–800. doi:10.1016/j.cej.2011.02.012 |
| 33. | Koech, P. K.; Zhang, J.; Kutnyakov, I. V.; Cosimbescu, L.; Lee, S.-J.; Bowden, M. E.; Smurthwaite, T. D.; Heldebrant, D. J. RSC Adv. 2013, 3, 566–572. doi:10.1039/C2RA22801G |
| 23. | Heldebrant, D. J.; Jessop, P. G.; Thomas, C. A.; Eckert, C. A.; Liotta, C. L. J. Org. Chem. 2005, 70, 5335–5338. doi:10.1021/jo0503759 |
| 39. | Yoshida, Y.; Aoyagi, N.; Endo, T. New J. Chem. 2017, 41, 14390–14396. doi:10.1039/C7NJ03133E |
© 2018 Yoshida et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)