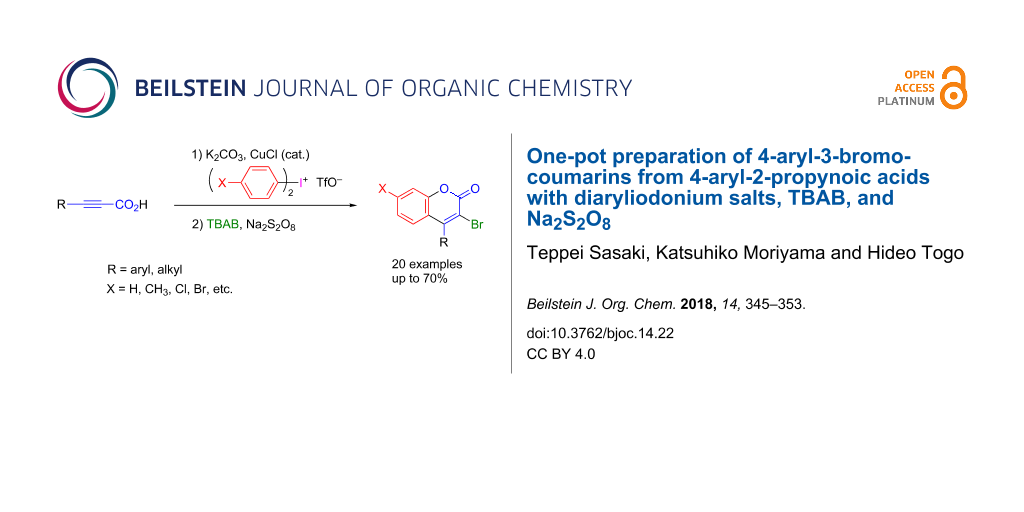
Abstract
Various 4-aryl-3-bromocoumarins were smoothly obtained in moderate yields in one pot by treating 3-aryl-2-propynoic acids with diaryliodonium triflates and K2CO3 in the presence of CuCl, followed by the reaction with tetrabutylammonium bromide (TBAB) and Na2S2O8. The obtained 3-bromo-4-phenylcoumarin was transformed into 4-phenylcoumarin derivatives bearing C–H, C–S, C–N, and C–C bonds at 3-position.

Graphical Abstract
Introduction
Coumarin is a benzo-α-pyrone and one of the typical heterocyclic compounds. The importance of coumarins arises from the fact that the coumarin skeleton is present in many natural products extracted from plants [1-3] and some of them show potent pharmacological activities, such as antidepressant [4], antimicrobial [5,6], antioxidants [7,8], anti-inflammatory [9,10], antinociceptive [11], antitumor [1], antiasthmatic [12], and antiviral including anti-HIV [13,14].
Comprehensive synthetic studies of coumarins and their derivatives have been carried out [15,16]. Typically, coumarins are prepared by the acid-catalyzed condensation of 2-alkynoic acids and phenols or the condensation of β-ketoesters and phenols (the Pechmann condensation) [17]. Recent studies for the metal-catalyzed reactions for the synthesis of the coumarin skeleton are as follows: the Yb(OTf)3-catalyzed microwave irradiation of phenols and propynoic acids [18], the Pd(OAc)2-catalyzed oxidative cyclocarbonylation of 2-vinylphenols at 110 °C [19], the FeCl3-catalyzed areneselenyl-cyclization of aryl 2-alkynoates with ArSeSeAr at rt [20], and the Rh-catalyzed annulation of arylthiocarbamates with alkynes/AgOTf/Cu(OAc)2 at 120 °C [21]. As examples of the transition-metal-free construction of the coumarin skeleton, the Brønsted acid-catalyzed reaction of phenols and propynoic acids [22] and the (−)-riboflavin-catalyzed photochemical reaction of cinnamic acids [23] were reported recently. Moreover, the use of radical cyclization for the construction of the coumarin skeleton has become widespread. Examples include the radical addition–cyclization reactions of aryl 2-alkynoates with RC(=O)CO2H/AgNO3(cat.)/K2S2O8 at 60 °C [24], with Cu(OAc)2/1-trifluoromethyl-3,3-dimethyl-1,2-benziodoxole (Togni reagent) at 60 °C [25], with R2P(=O)H/Ag2CO3(cat.)/Mg(NO3)2 at 100 °C [26], with BrCF2CO2Et/fac-Ir(ppy)3(cat.) under irradiation at rt [27], with R-CH=O/(n-Bu)4NBr (TBAB, cat.)/K2S2O8 at 90 °C [28], with ArSO2H/Eosin Y(cat.)/tert-butyl hydrogen peroxide (TBHP) at rt [29], and with ArSO2NHNH2/n-Bu4NI(cat.)/TBHP at 80 °C [30].
In addition, the formation of coumarins via the bromine-radical-mediated reaction of aryl 2-alkynoates with TBAB/K2S2O8 at 90 °C [31], the cyanomethyl-radical-mediated reaction of aryl 2-alkynoates with tert-butyl peroxybenzoate (TBPB)/acetonitrile at 130 °C [32], the sunlight-promoted reaction of aryl 2-alkynoates with N-iodosuccinimide (NIS) at rt [33], and the visible-light-mediated reaction of aryl 2-alkynoates with N-bromosuccinimide (NBS) at rt [34], where those reactions proceed via radical spiro-cyclization and then radical 1,2-carboxyl group migration, were reported.
On the other hand, diaryliodonium salts are very useful for the C-arylation of active CH groups, the O-arylation of OH groups, and the N-arylation of NH groups under metal-free conditions [35-39]. For example, treatment of arenecarboxylic acids and alkanecarboxylic acids with diaryliodonium salts and t-BuOK under toluene refluxing conditions provides the corresponding aryl carboxylates in good yields [40,41]. However, the O-arylation of 2-alkynoic acids, which are much more acidic than arenecarboxylic acids and alkanecarboxylic acids, and therefore, the conjugate bases of 2-alkynoic acids are much less nucleophilic than those of arenecarboxylic acids and alkanecarboxylic acids, was not studied. On the other hand, it is known that 4-arylcoumarins have antitumor activity [42]. Therefore, the one-pot preparation of 4-arylcoumarins from 3-aryl-2-alkynoic acids via aryl esters and cyclization is attractive and important.
Here, as part of our ongoing investigation of the synthetic use of diaryliodonium salts for the preparation of heterocyclic compounds [43-46], we would like to report an efficient one-pot preparation of 4-aryl-3-bromocoumarins by treatment of 3-aryl-2-propynoic acids with diaryliodonium triflate in the presence of a base, followed by the reaction with tetrabutylammonium bromide (TBAB) and Na2S2O8 in a mixture of 1,2-dichloroethane and water [31].
Results and Discussion
First, treatment of 3-phenyl-2-propynoic acid (1a, 0.5 mmol) with diphenyliodonium triflate (A, 1.0 equiv) in the presence of CuCl (5 mol %) and K2CO3 (1.0 equiv) in dichloromethane (3.0 mL) at 40 °C based on a previous report [46] gave phenyl 3-phenyl-2-propynoate (2Aa) in 46% yield, as shown in Table 1, entry 1. When the amount of the solvent was increased to 7.5 mL under the same conditions, the yield of phenyl ester 2Aa was increased to 74% (Table 1, entry 2). Under the same conditions, the base was changed to NaH, Cs2CO3, t-BuOK, NaNH2, and K3PO4 instead of K2CO3. However, the yield of phenyl ester 2Aa was moderate to low (Table 1, entries 3–7). When the amount of K2CO3 was reduced to 0.5 equiv under the same conditions as those in entry 2, the yield of phenyl ester 2Aa was increased to 80% (Table 1, entry 8). When CuCl was changed to CuI and CuBr, the difference of the yield of phenyl ester 2Aa was small, but CuCl gave the highest yield (Table 1, entries 8–10). Then, the reaction temperature was changed to 0 °C, rt, 50 °C, and 60 °C under the same conditions as those in entry 8, and phenyl ester 2Aa was obtained in 83% yield at 50 °C (Table 1, entries 11–14). On the other hand, when the present reaction was carried out without CuCl under the same conditions, phenyl ester 2Aa was not obtained at all (Table 1, entry 15).
Table 1: O-Phenylation of 3-phenyl-2-propynoic acid (1a) with diphenyliodonium triflate (A).
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-22-i5.svg?max-width=637&scale=1.0)
|
|||||
| entry | base | solvent (mL) | additive (mol %) | temp. (°C) | yield (%) |
| 1 | K2CO3 (1.0) | CH2Cl2 (3.0) | CuCl (5) | 40 | 46 |
| 2 | K2CO3 (1.0) | CH2Cl2 (7.5) | CuCl (5) | 40 | 74 |
| 3 | NaH (1.0) | CH2Cl2 (7.5) | CuCl (5) | 40 | 24 |
| 4 | Cs2CO3 (0.5) | CH2Cl2 (7.5) | CuCl (5) | 40 | 17 |
| 5 | t-BuOK (1.0) | CH2Cl2 (7.5) | CuCl (5) | 40 | 48 |
| 6 | NaNH2 (1.0) | CH2Cl2 (7.5) | CuCl (5) | 40 | 9 |
| 7 | K3PO4 (1.0) | CH2Cl2 (7.5) | CuCl (5) | 40 | 30 |
| 8 | K2CO3 (0.5) | CH2Cl2 (7.5) | CuCl (5) | 40 | 80 |
| 9 | K2CO3 (0.5) | CH2Cl2 (7.5) | CuI (5) | 40 | 78 |
| 10 | K2CO3 (0.5) | CH2Cl2 (7.5) | CuBr (5) | 40 | 77 |
| 11 | K2CO3 (0.5) | CH2Cl2 (7.5) | CuCl (5) | 0 | 11 |
| 12 | K2CO3 (0.5) | CH2Cl2 (7.5) | CuCl (5) | rt | 71 |
| 13 | K2CO3 (0.5) | DCE (7.5) | CuCl (5) | 50 | 83 |
| 14 | K2CO3 (0.5) | DCE (7.5) | CuCl (5) | 60 | 75 |
| 15 | K2CO3 (0.5) | DCE (7.5) | – | 50 | 0 |
Then, the iodocyclization of phenyl ester 2Aa to 3-iodo-4-phenylcoumarin (3Aa’) with N-iodosuccinimide (NIS, 2.0 equiv)/BF3·Et2O (2.0 or 1.1 equiv) was studied based on the previous reports [45,46], as shown in Table 2. However, 3-iodo-4-phenylcoumarin (3Aa’) was obtained in low to moderate yields (Table 2, entries 1 and 2). To improve the yield of 3-halo-4-phenylcoumarins 3Aa or 3Aa’, the halocyclization of 2Aa with N-bromosuccinimide (NBS, 2.0 equiv)/BF3·Et2O (1.1 equiv), with 1,3-diiodo-5,5-dimethylhydantoin (DIH, 2.0 equiv)/BF3·Et2O (1.1 equiv), and with 1,3-dibromo-5,5-dimethylhydantoin (DBH, 2.0 equiv)/BF3·Et2O (1.1 equiv) was carried out to form 3-bromo-4-phenylcoumarin (3Aa), 3-iodo-4-phenylcoumarin (3Aa’), and 3-bromo-4-phenylcoumarin (3Aa) in 28, 49 and 46% yields, respectively (Table 2, entries 3–5). The treatment of phenyl ester 2Aa with molecular iodine (2.0 equiv)/K2CO3 (2.0 equiv) did not generate 3-iodo-4-phenylcoumarin (3Aa’) at all (Table 2, entry 6). Thus, the iodonium-based or bromonium-based electrophilic cyclization of phenyl 3-phenyl-2-propynoate (2Aa) does not proceed efficiently. Then, the bromo-radical-based cyclization of phenyl 3-phenyl-2-propynoate (2Aa) with tetrabutylammonium bromide (TBAB, 2.0 equiv)/Na2S2O8 (1.5 equiv) [31] in a mixture of 1,2-dichloroethane (DCE) and water at 90 °C was carried out to give 3-bromo-4-phenylcoumarin (3Aa) in 68% yield (Table 2, entry 7). When the iodocyclization of phenyl ester 2Aa with tetrabutylammonium iodide (TBAI, 2.0 equiv)/Na2S2O8 (1.5 equiv) was carried out, the yield of iodocyclization product 3Aa’ was decreased to 45% (Table 2, entry 8). When the bromocyclization of phenyl ester 2Aa with TBAB (2.0 equiv)/Na2S2O8 (1.0 equiv) in a mixture of DCE and water at 90 °C was carried out, 3-bromo-4-phenylcoumarin (3Aa) was obtained in 79% yield (Table 2, entry 9). When Na2S2O8 was increased to 2.0 equivalents or TBAB was increased to 2.5 equivalents under the same conditions, the yields of 3-bromo-4-phenylcoumarin (3Aa) were decreased to 51 and 69%, respectively (Table 2, entries 10 and 11). Moreover, when Na2S2O8 was changed to K2S2O8, (NH4)2S2O8, and Oxone® (2KHSO5·KHSO4·K2SO4), the yields of 3-bromo-4-phenylcoumarin (3Aa) were decreased to 71, 69 and 37%, respectively (Table 2, entries 12–14). Thus, it was confirmed that the treatment of phenyl ester 2Aa with TBAB (2.0 equiv)/Na2S2O8 (1.0 equiv) in a mixture of DCE and water at 90 °C for 19 h was the most efficient, giving 3-bromo-4-phenylcoumarin (3Aa) in good yield (Table 2, entry 9).
Table 2: Halocyclization of phenyl 3-phenyl-2-propynoate (2Aa) to 3-halo-4-phenylcoumarins 3Aa and 3Aa’.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-22-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | additive (equiv) | solvent (mL) | temp. (°C) | time (h) | yield (%) |
| 1 | NIS (2.0), BF3·Et2O (2.0) | CH2Cl2 (3.0) | 40 | 1 | 36 (3Aa’) |
| 2 | NIS (2.0), BF3·Et2O (1.1) | CH2Cl2 (3.0) | 40 | 1 | 45 (3Aa’) |
| 3 | NBS (2.0), BF3·Et2O (1.1) | CH2Cl2 (3.0) | 40 | 1 | 28 (3Aa) |
| 4 | DIH (2.0), BF3·Et2O (1.1) | CH2Cl2 (3.0) | 40 | 1 | 49 (3Aa’) |
| 5 | DBH (2.0), BF3·Et2O (1.1) | CH2Cl2 (3.0) | 40 | 1 | 46 (3Aa) |
| 6 | I2 (2.0), K2CO3 (2.0) | CH3CN (3.0) | 40 | 1 | 0 |
| 7 | TBAB (2.0), Na2S2O8 (1.5) | DCE:H2O (1:1, 5.0) | 90 | 19 | 68 (3Aa) |
| 8 | TBAI (2.0), Na2S2O8 (1.5) | DCE:H2O (1:1, 5.0) | 90 | 19 | 45 (3Aa’) |
| 9 | TBAB (2.0), Na2S2O8 (1.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 79 (3Aa) |
| 10 | TBAB (2.0), Na2S2O8 (2.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 51 (3Aa) |
| 11 | TBAB (2.5), Na2S2O8 (1.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 69 (3Aa) |
| 12 | TBAB (2.0), K2S2O8 (1.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 71 (3Aa) |
| 13 | TBAB (2.0), (NH4)2S2O8 (1.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 69 (3Aa) |
| 14 | TBAB (2.0), Oxone® (1.0) | DCE:H2O (1:1, 5.0) | 90 | 19 | 37 (3Aa) |
Finally, based on the results in Table 1 and Table 2, the one-pot preparation of 4-aryl-3-bromocoumarins 3 from 3-aryl-2-propynoic acids 1 was carried out. 3-Aryl-2-propynoic acids 1, such as 3-phenyl-2-propynoic acid (1a), 3-(o-methylphenyl)-2-propynoic acid (1b), 3-(m-methylphenyl)-2-propynoic acid (1c), 3-(p-methylphenyl)-2-propynoic acid (1d), 3-(p-methoxyphenyl)-2-propynoic acid (1e), 3-(p-fluorophenyl)-2-propynoic acid (1f), 3-(p-chlorophenyl)-2-propynoic acid (1g), 3-(o-chlorophenyl)-2-propynoic acid (1h), 3-(m-chlorophenyl)-2-propynoic acid (1i), 3-(p-bromophenyl)-2-propynoic acid (1j), 3-(p-biphenyl)-2-propynoic acid (1k), 3-(naphthalen-2’-yl)-2-propynoic acid (1l), and 3-(naphthalen-1’-yl)-2-propynoic acid (1m), were treated with diphenyliodonium triflate (A, 1.0 equiv) in the presence of CuCl and K2CO3 in CH2Cl2 for 3 h under refluxing conditions. After removal of the solvent, the second-step treatment of the reaction mixture with TBAB (2.0 equiv) and Na2S2O8 (2.0 equiv) in a mixture of DCE and water at 90 °C for 19 h gave 4-aryl-3-bromocoumarins 3Aa–3Am in moderate yields, respectively, as shown in Scheme 1. As a gram-scale experiment, treatment of 3-phenyl-2-propynoic acid (1a, 8 mmol) with diphenyliodonium triflate A in the presence of CuCl and K2CO3 in CH2Cl2 for 3 h, followed by removal of the solvent and the reaction with TBAB and Na2S2O8 in a mixture of DCE and water at 90 °C for 19 h gave 3-bromo-4-phenylcoumarin (3Aa) in 52% yield. For 3-aryl-2-propynoic acids bearing heteroaromatic groups, treatment of 3-(benzothiophen-2’-yl)-2-propynoic acid (1n) and 3-(benzofuran-2’-yl)-2-propynoic acid (1o) under the same procedure and conditions gave the corresponding coumarins 3An and 3Ao in moderate yields, respectively. Under the present procedure and conditions, use of 2-hexynoic acid (1p), a 3-alkyl-2-propynoic acid, provided 3-bromo-4-propylcoumarin (3Ap) in 42% yield, as shown in Scheme 1.
![[1860-5397-14-22-i1]](/bjoc/content/inline/1860-5397-14-22-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot preparation of 4-aryl-3-bromocoumarins 3 from 3-aryl-2-propynoic acids 1 with diphenyliodonium triflate (A). a3-Phenyl-2-propynoic acid (1a, 8.0 mmol) was used. bThe first reaction step was conducted with K2CO3 (1.0 equiv) under refluxing conditions.
Scheme 1: One-pot preparation of 4-aryl-3-bromocoumarins 3 from 3-aryl-2-propynoic acids 1 with diphenyliodon...
Then, other diaryliodonium triflates were used instead of diphenyliodonium triflate (A). Treatment of 3-phenyl-2-propynoic acid (1a) with diaryliodonium triflates (1.0 equiv), such as di(p-methylphenyl)iodonium triflate (B), di(tert-butylphenyl)iodonium triflate (C), di(p-chlorophenyl)iodonium triflate (D), and di(p-bromophenyl)iodonium triflate (E), in the presence of CuCl and K2CO3 in CH2Cl2 for 3 h under refluxing conditions, followed by removal of the solvent and the reaction with TBAB (2.0 equiv) and Na2S2O8 (2.0 equiv) in a mixture of DCE and water at 90 °C for 19 h gave 3-bromo-4-phenylcoumarin derivatives 3Ba–3Ea bearing methyl, tert-butyl, chloro, and bromo groups at 7-position in good to moderate yields, respectively, as shown in Scheme 2.
![[1860-5397-14-22-i2]](/bjoc/content/inline/1860-5397-14-22-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: One-pot preparation of 3-bromo-4-phenylcoumarins 3a from 3-phenyl-2-propynoic acid (1a) with daryliodonium triflates B–E.
Scheme 2: One-pot preparation of 3-bromo-4-phenylcoumarins 3a from 3-phenyl-2-propynoic acid (1a) with daryli...
As regards the synthetic utilization of the products in the present one-pot reaction, treatment of 3-bromo-4-phenylcoumarin (3Aa) with Zn in ethanol under refluxing conditions gave 4-phenylcoumarin (4Aa) in 81% yield. Treatment of 3-bromo-4-phenylcoumarin (3Aa) with p-toluenethiol/N,N’-dimethylethylenediamine (DMEDA)/K2CO3 in the presence of CuI in toluene at refluxing temperature and with p-methoxybenzamide/DMEDA/K2CO3 in the presence of CuI in toluene at refluxing temperature generated 3-(4-methylbenzenesulfenyl)-4-phenylcoumarin (5Aa) and 3-(4-methoxylbenzoylamino)-4-phenylcoumarin (6Aa) in 62 and 51% yields, respectively. The Pd-catalyzed coupling reactions of 3-bromo-4-phenylcoumarin (3Aa) with 4-methylstyrene/K2CO3/PdCl2(Ph3P)2, with phenylacetylene/PdCl2(Ph3P)2/Et3N and with PhB(OH)2/K2CO3/PdCl2(Ph3P)2 provided the corresponding C–C bonded coumarin derivatives 7Aa, 8Aa, and 9Aa in 79, 60 and 76% yields, respectively (Scheme 3).
![[1860-5397-14-22-i3]](/bjoc/content/inline/1860-5397-14-22-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Derivatization of 3-bromo-4-phenylcoumarin.
Scheme 3: Derivatization of 3-bromo-4-phenylcoumarin.
To support the present bromocyclization reaction to form 4-aryl-3-bromocoumarins with TBAB and Na2S2O8 at the second step, the present one-pot preparation of 3-bromo-4-phenylcoumarin (3Aa) from 3-phenyl-2-propynoic acid (1a) was carried out in the presence of 2,2,6,6-tetramethylpiperidine 1-oxyl radical (TEMPO, 2.0 equiv) or 2,6-di(tert-butyl-p-cresol (BHT, 3.0 equiv) at the second step under the same procedure and conditions, but 3-bromo-4-phenylcoumarin (3Aa) was not obtained at all in both reactions. Thus, the present bromocyclization of the formed phenyl 3-phenyl-2-propynoate (2Aa) with TBAB and Na2S2O8 in a mixture of DCE and water is a radical-mediated bromocyclization reaction. X-ray crystallographic analysis of 3-bromo-7-chloro-4-phenylcoumarin (3Da), which was formed by the subsequent treatment of 3-phenyl-2-propynoic acid (1a) with di(p-chlorophenyl)iodonium triflate (D) and then with TBAB and Na2S2O8, was carried out, as shown in Figure 1. Based on those results, the possible reaction pathway is shown in Scheme 4.
![[1860-5397-14-22-1]](/bjoc/content/figures/1860-5397-14-22-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP of 3-bromo-7-chloro-4-phenylcoumarin (3Da).
Figure 1: ORTEP of 3-bromo-7-chloro-4-phenylcoumarin (3Da).
![[1860-5397-14-22-i4]](/bjoc/content/inline/1860-5397-14-22-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The O-arylation of 3-aryl-2-propynoic acid 1 with diaryliodonium triflate in the presence of K2CO3 and CuCl occurs to form aryl 3-aryl-2-propynoate 2 (1st step). The bromocyclization of aryl 3-aryl-2-propynoate 2 with TBAB and Na2S2O8 proceeds via a bromoradical addition to the triple bond to form very reactive vinyl radical I [47]. Ipso-cyclization of the vinyl radical I occurs to form spiro radical intermediate II. Then, β-cleavage of the spiro radical intermediate II proceeds to form carboxyl radical III. 6-Exo-trig cyclization of the carboxyl radical III onto the aromatic ring takes place to form adduct radical IV, which would be rapidly oxidized by Na2S2O8 to form cation intermediate V. Smooth deprotonation of cation intermediate V occurs to generate 4-aryl-3-bromocoumarin 3 (2nd step). The radical ipso-cyclization of the formed vinyl radical and its 1,2-carboxyl group migration agree with previously reported results [31-34].
Conclusion
The successive treatment of 3-aryl-2-propynoic acids with diaryliodonium triflates in the presence of K2CO3 and CuCl, and then with tetrabutylammonium bromide (TBAB) and Na2S2O8 gave 4-aryl-3-bromocoumarins bearing hydrogen, methyl, tert-butyl, chloro, and bromo groups at 7-position in moderate yields, respectively. In one of the obtained 4-aryl-3-bromocoumarins, the C–Br bond of 3-bromo-4-phenylcoumarin was smoothly converted into 4-phenylcoumarins bearing C–H, C–S, C–N, and C–C bonds at 3-position. We believe the present method will be useful for the preparation of various 4-arylcoumarin derivatives due to its simple one-pot synthesis.
Supporting Information
| Supporting Information File 1: NMR charts of all coumarin derivatives 3Aa–3Ap, 3Ba–3Ea, and 4Aa–9Aa, and X-ray analytical data of 3Da. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Lacy, A. Curr. Pharm. Des. 2004, 10, 3797–3811. doi:10.2174/1381612043382693
Return to citation in text: [1] [2] -
Musa, M. A.; Badisa, V. L. D.; Latinwo, L. M.; Waryoba, C.; Ugochukwu, N. Anticancer Res. 2010, 30, 4613–4617.
Return to citation in text: [1] -
Medina, F. G.; Marrero, J. G.; Macías-Alonso, M.; González, M. C.; Córdova-Guerrero, I.; García, A. G. T.; Osegueda-Robles, S. Nat. Prod. Rep. 2015, 32, 1472–1507. doi:10.1039/C4NP00162A
Return to citation in text: [1] -
Sashidhara, K. V.; Kumar, A.; Chatterjee, M.; Rao, K. B.; Singh, S.; Verma, A. K.; Palit, G. Bioorg. Med. Chem. Lett. 2011, 21, 1937–1941. doi:10.1016/j.bmcl.2011.02.040
Return to citation in text: [1] -
Ostrov, D. A.; Hernández-Prada, J. A.; Corsino, P. E.; Finton, K. A.; Le, N.; Rowe, T. C. Antimicrob. Agents Chemother. 2007, 51, 3688–3698. doi:10.1128/AAC.00392-07
Return to citation in text: [1] -
Chimenti, F.; Bizzarri, B.; Bolasco, A.; Secci, D.; Chimenti, P.; Granese, A.; Carradori, S.; Rivanera, D.; Zicari, A.; Scaltrito, M. M.; Sisto, F. Bioorg. Med. Chem. Lett. 2010, 20, 4922–4926. doi:10.1016/j.bmcl.2010.06.048
Return to citation in text: [1] -
Kostova, I.; Bhatia, S.; Grigorov, P.; Balkansky, S.; Parmar, V. S.; Prasad, A. K.; Saso, L. Curr. Med. Chem. 2011, 18, 3929–3951. doi:10.2174/092986711803414395
Return to citation in text: [1] -
Xi, G.-L.; Liu, Z.-Q. J. Agric. Food Chem. 2015, 63, 3516–3523. doi:10.1021/acs.jafc.5b00399
Return to citation in text: [1] -
Bansal, Y.; Sethi, P.; Bansal, G. Med. Chem. Res. 2013, 22, 3049–3060. doi:10.1007/s00044-012-0321-6
Return to citation in text: [1] -
Fylaktakidou, K. C.; Hadjipavlou-Litina, D. J.; Litinas, K. E.; Nicolaides, D. N. Curr. Pharm. Des. 2004, 10, 3813–3833. doi:10.2174/1381612043382710
Return to citation in text: [1] -
De Almeida Barros, T. A.; De Freitas, L. A. R.; Filho, J. M. B.; Nunes, X. P.; Giulietti, A. M.; De Souza, G. E.; Dos Santos, R. R.; Soares, M. B. P.; Villarreal, C. F. J. Pharm. Pharmacol. 2010, 62, 205–213. doi:10.1211/jpp.62.02.0008
Return to citation in text: [1] -
Sánchez-Recillas, A.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Rios, M. Y.; Ibarra-Barajas, M.; Estrada-Soto, S. Eur. J. Med. Chem. 2014, 77, 400–408. doi:10.1016/j.ejmech.2014.03.029
Return to citation in text: [1] -
Hwu, J. R.; Lin, S.-Y.; Tsay, S.-C.; De Clercq, E.; Leyssen, P.; Neyts, J. J. Med. Chem. 2011, 54, 2114–2126. doi:10.1021/jm101337v
Return to citation in text: [1] -
Ong, E. B. B.; Watanabe, N.; Saito, A.; Futamura, Y.; El Galil, K. H. A.; Koito, A.; Najimudin, N.; Osada, H. J. Biol. Chem. 2011, 286, 14049–14056. doi:10.1074/jbc.M110.185397
Return to citation in text: [1] -
Vekariya, R. H.; Patel, H. D. Synth. Commun. 2014, 44, 2756–2788. doi:10.1080/00397911.2014.926374
Return to citation in text: [1] -
Žalubovskis, R. Chem. Heterocycl. Compd. (Engl. Transl.) 2015, 51, 607–612. doi:10.1007/s10593-015-1748-8
Return to citation in text: [1] -
Sethna, S.; Phadke, R. Org. React. 2011, 7, 1–58. doi:10.1002/0471264180.or007.01
Return to citation in text: [1] -
Fiorito, S.; Epifano, F.; Taddeo, V. A.; Genovese, S. Tetrahedron Lett. 2016, 57, 2939–2942. doi:10.1016/j.tetlet.2016.05.087
Return to citation in text: [1] -
Ferguson, J.; Zeng, F.; Alper, H. Org. Lett. 2012, 14, 5602–5605. doi:10.1021/ol302725x
Return to citation in text: [1] -
Mantovani, A. C.; Goulart, T. A. C.; Back, D. F.; Menezes, P. H.; Zeni, G. J. Org. Chem. 2014, 79, 10526–10536. doi:10.1021/jo502199q
Return to citation in text: [1] -
Zhao, Y.; Han, F.; Yang, L.; Xia, C. Org. Lett. 2015, 17, 1477–1480. doi:10.1021/acs.orglett.5b00364
Return to citation in text: [1] -
Choi, H.; Kim, J.; Lee, K. Tetrahedron Lett. 2016, 57, 3600–3603. doi:10.1016/j.tetlet.2016.06.039
Return to citation in text: [1] -
Metternich, J. B.; Gilmour, R. J. Am. Chem. Soc. 2016, 138, 1040–1045. doi:10.1021/jacs.5b12081
Return to citation in text: [1] -
Yan, K.; Yang, D.; Wei, W.; Wang, F.; Shuai, Y.; Li, Q.; Wang, H. J. Org. Chem. 2015, 80, 1550–1556. doi:10.1021/jo502474z
Return to citation in text: [1] -
Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m
Return to citation in text: [1] -
Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Org. Lett. 2014, 16, 3356–3359. doi:10.1021/ol5013839
Return to citation in text: [1] -
Fu, W.; Zhu, M.; Zou, G.; Xu, C.; Wang, Z.; Ji, B. J. Org. Chem. 2015, 80, 4766–4770. doi:10.1021/acs.joc.5b00305
Return to citation in text: [1] -
Mi, X.; Wang, C.; Huang, M.; Wu, Y.; Wu, Y. J. Org. Chem. 2015, 80, 148–155. doi:10.1021/jo502220b
Return to citation in text: [1] -
Yang, W.; Yang, S.; Li, P.; Wang, L. Chem. Commun. 2015, 51, 7520–7523. doi:10.1039/C5CC00878F
Return to citation in text: [1] -
Wei, W.; Wen, J.; Yang, D.; Guo, M.; Wang, Y.; You, J.; Wang, H. Chem. Commun. 2015, 51, 768–771. doi:10.1039/C4CC08117J
Return to citation in text: [1] -
Qiu, G.; Liu, T.; Ding, Q. Org. Chem. Front. 2016, 3, 510–515. doi:10.1039/C6QO00041J
Return to citation in text: [1] [2] [3] [4] -
Yu, Y.; Zhuang, S.; Liu, P.; Sun, P. J. Org. Chem. 2016, 81, 11489–11495. doi:10.1021/acs.joc.6b02155
Return to citation in text: [1] [2] -
Ni, S.; Cao, J.; Mei, H.; Han, J.; Li, S.; Pan, Y. Green Chem. 2016, 18, 3935–3939. doi:10.1039/C6GC01027J
Return to citation in text: [1] [2] -
Feng, S.; Li, J.; Li, Z.; Sun, H.; Shi, H.; Wang, X.; Xie, X.; She, X. Org. Biomol. Chem. 2017, 15, 8820–8826. doi:10.1039/C7OB02199B
Return to citation in text: [1] [2] -
Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Olofsson, B. Top. Curr. Chem. 2015, 373, 135–166. doi:10.1007/128_2015_661
Return to citation in text: [1] -
Fañanás-Mastral, M. Synthesis 2017, 49, 1905–1930. doi:10.1055/s-0036-1589483
Return to citation in text: [1] -
Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732
Return to citation in text: [1] -
Cao, C. K.; Sheng, J.; Chen, C. Synthesis 2017, 49, 5081–5092. doi:10.1055/s-0036-1589515
Return to citation in text: [1] -
Petersen, T. B.; Khan, R.; Olofsson, B. Org. Lett. 2011, 13, 3462–3465. doi:10.1021/ol2012082
Return to citation in text: [1] -
Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645
Return to citation in text: [1] -
Mutai, P.; Breuzard, G.; Pagano, A.; Allegro, D.; Peyrot, V.; Chibale, K. Bioorg. Med. Chem. 2017, 25, 1652–1665. doi:10.1016/j.bmc.2017.01.035
Return to citation in text: [1] -
Kakinuma, Y.; Moriyama, K.; Togo, H. Synthesis 2013, 45, 183–188. doi:10.1055/s-0032-1316824
Return to citation in text: [1] -
Miyagi, K.; Moriyama, K.; Togo, H. Heterocycles 2014, 89, 2122–2136. doi:10.3987/COM-14-13071
Return to citation in text: [1] -
Sasaki, T.; Miyagi, K.; Moriyama, K.; Togo, H. Org. Lett. 2016, 18, 944–947. doi:10.1021/acs.orglett.5b03651
Return to citation in text: [1] [2] -
Sasaki, T.; Moriyama, K.; Togo, H. J. Org. Chem. 2017, 82, 11727–11734. doi:10.1021/acs.joc.7b01433
Return to citation in text: [1] [2] [3] -
Togo, H. Advanced Free Radicals for Organic Synthesis; Chapter 3; Elsevier: Oxford, 2004; pp 75–94.
Return to citation in text: [1]
| 34. | Feng, S.; Li, J.; Li, Z.; Sun, H.; Shi, H.; Wang, X.; Xie, X.; She, X. Org. Biomol. Chem. 2017, 15, 8820–8826. doi:10.1039/C7OB02199B |
| 35. | Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 36. | Olofsson, B. Top. Curr. Chem. 2015, 373, 135–166. doi:10.1007/128_2015_661 |
| 37. | Fañanás-Mastral, M. Synthesis 2017, 49, 1905–1930. doi:10.1055/s-0036-1589483 |
| 38. | Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732 |
| 39. | Cao, C. K.; Sheng, J.; Chen, C. Synthesis 2017, 49, 5081–5092. doi:10.1055/s-0036-1589515 |
| 40. | Petersen, T. B.; Khan, R.; Olofsson, B. Org. Lett. 2011, 13, 3462–3465. doi:10.1021/ol2012082 |
| 41. | Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645 |
| 1. | Lacy, A. Curr. Pharm. Des. 2004, 10, 3797–3811. doi:10.2174/1381612043382693 |
| 2. | Musa, M. A.; Badisa, V. L. D.; Latinwo, L. M.; Waryoba, C.; Ugochukwu, N. Anticancer Res. 2010, 30, 4613–4617. |
| 3. | Medina, F. G.; Marrero, J. G.; Macías-Alonso, M.; González, M. C.; Córdova-Guerrero, I.; García, A. G. T.; Osegueda-Robles, S. Nat. Prod. Rep. 2015, 32, 1472–1507. doi:10.1039/C4NP00162A |
| 9. | Bansal, Y.; Sethi, P.; Bansal, G. Med. Chem. Res. 2013, 22, 3049–3060. doi:10.1007/s00044-012-0321-6 |
| 10. | Fylaktakidou, K. C.; Hadjipavlou-Litina, D. J.; Litinas, K. E.; Nicolaides, D. N. Curr. Pharm. Des. 2004, 10, 3813–3833. doi:10.2174/1381612043382710 |
| 21. | Zhao, Y.; Han, F.; Yang, L.; Xia, C. Org. Lett. 2015, 17, 1477–1480. doi:10.1021/acs.orglett.5b00364 |
| 47. | Togo, H. Advanced Free Radicals for Organic Synthesis; Chapter 3; Elsevier: Oxford, 2004; pp 75–94. |
| 7. | Kostova, I.; Bhatia, S.; Grigorov, P.; Balkansky, S.; Parmar, V. S.; Prasad, A. K.; Saso, L. Curr. Med. Chem. 2011, 18, 3929–3951. doi:10.2174/092986711803414395 |
| 8. | Xi, G.-L.; Liu, Z.-Q. J. Agric. Food Chem. 2015, 63, 3516–3523. doi:10.1021/acs.jafc.5b00399 |
| 22. | Choi, H.; Kim, J.; Lee, K. Tetrahedron Lett. 2016, 57, 3600–3603. doi:10.1016/j.tetlet.2016.06.039 |
| 31. | Qiu, G.; Liu, T.; Ding, Q. Org. Chem. Front. 2016, 3, 510–515. doi:10.1039/C6QO00041J |
| 32. | Yu, Y.; Zhuang, S.; Liu, P.; Sun, P. J. Org. Chem. 2016, 81, 11489–11495. doi:10.1021/acs.joc.6b02155 |
| 33. | Ni, S.; Cao, J.; Mei, H.; Han, J.; Li, S.; Pan, Y. Green Chem. 2016, 18, 3935–3939. doi:10.1039/C6GC01027J |
| 34. | Feng, S.; Li, J.; Li, Z.; Sun, H.; Shi, H.; Wang, X.; Xie, X.; She, X. Org. Biomol. Chem. 2017, 15, 8820–8826. doi:10.1039/C7OB02199B |
| 5. | Ostrov, D. A.; Hernández-Prada, J. A.; Corsino, P. E.; Finton, K. A.; Le, N.; Rowe, T. C. Antimicrob. Agents Chemother. 2007, 51, 3688–3698. doi:10.1128/AAC.00392-07 |
| 6. | Chimenti, F.; Bizzarri, B.; Bolasco, A.; Secci, D.; Chimenti, P.; Granese, A.; Carradori, S.; Rivanera, D.; Zicari, A.; Scaltrito, M. M.; Sisto, F. Bioorg. Med. Chem. Lett. 2010, 20, 4922–4926. doi:10.1016/j.bmcl.2010.06.048 |
| 19. | Ferguson, J.; Zeng, F.; Alper, H. Org. Lett. 2012, 14, 5602–5605. doi:10.1021/ol302725x |
| 45. | Sasaki, T.; Miyagi, K.; Moriyama, K.; Togo, H. Org. Lett. 2016, 18, 944–947. doi:10.1021/acs.orglett.5b03651 |
| 46. | Sasaki, T.; Moriyama, K.; Togo, H. J. Org. Chem. 2017, 82, 11727–11734. doi:10.1021/acs.joc.7b01433 |
| 4. | Sashidhara, K. V.; Kumar, A.; Chatterjee, M.; Rao, K. B.; Singh, S.; Verma, A. K.; Palit, G. Bioorg. Med. Chem. Lett. 2011, 21, 1937–1941. doi:10.1016/j.bmcl.2011.02.040 |
| 20. | Mantovani, A. C.; Goulart, T. A. C.; Back, D. F.; Menezes, P. H.; Zeni, G. J. Org. Chem. 2014, 79, 10526–10536. doi:10.1021/jo502199q |
| 31. | Qiu, G.; Liu, T.; Ding, Q. Org. Chem. Front. 2016, 3, 510–515. doi:10.1039/C6QO00041J |
| 13. | Hwu, J. R.; Lin, S.-Y.; Tsay, S.-C.; De Clercq, E.; Leyssen, P.; Neyts, J. J. Med. Chem. 2011, 54, 2114–2126. doi:10.1021/jm101337v |
| 14. | Ong, E. B. B.; Watanabe, N.; Saito, A.; Futamura, Y.; El Galil, K. H. A.; Koito, A.; Najimudin, N.; Osada, H. J. Biol. Chem. 2011, 286, 14049–14056. doi:10.1074/jbc.M110.185397 |
| 17. | Sethna, S.; Phadke, R. Org. React. 2011, 7, 1–58. doi:10.1002/0471264180.or007.01 |
| 31. | Qiu, G.; Liu, T.; Ding, Q. Org. Chem. Front. 2016, 3, 510–515. doi:10.1039/C6QO00041J |
| 12. | Sánchez-Recillas, A.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Rios, M. Y.; Ibarra-Barajas, M.; Estrada-Soto, S. Eur. J. Med. Chem. 2014, 77, 400–408. doi:10.1016/j.ejmech.2014.03.029 |
| 18. | Fiorito, S.; Epifano, F.; Taddeo, V. A.; Genovese, S. Tetrahedron Lett. 2016, 57, 2939–2942. doi:10.1016/j.tetlet.2016.05.087 |
| 46. | Sasaki, T.; Moriyama, K.; Togo, H. J. Org. Chem. 2017, 82, 11727–11734. doi:10.1021/acs.joc.7b01433 |
| 42. | Mutai, P.; Breuzard, G.; Pagano, A.; Allegro, D.; Peyrot, V.; Chibale, K. Bioorg. Med. Chem. 2017, 25, 1652–1665. doi:10.1016/j.bmc.2017.01.035 |
| 11. | De Almeida Barros, T. A.; De Freitas, L. A. R.; Filho, J. M. B.; Nunes, X. P.; Giulietti, A. M.; De Souza, G. E.; Dos Santos, R. R.; Soares, M. B. P.; Villarreal, C. F. J. Pharm. Pharmacol. 2010, 62, 205–213. doi:10.1211/jpp.62.02.0008 |
| 15. | Vekariya, R. H.; Patel, H. D. Synth. Commun. 2014, 44, 2756–2788. doi:10.1080/00397911.2014.926374 |
| 16. | Žalubovskis, R. Chem. Heterocycl. Compd. (Engl. Transl.) 2015, 51, 607–612. doi:10.1007/s10593-015-1748-8 |
| 43. | Kakinuma, Y.; Moriyama, K.; Togo, H. Synthesis 2013, 45, 183–188. doi:10.1055/s-0032-1316824 |
| 44. | Miyagi, K.; Moriyama, K.; Togo, H. Heterocycles 2014, 89, 2122–2136. doi:10.3987/COM-14-13071 |
| 45. | Sasaki, T.; Miyagi, K.; Moriyama, K.; Togo, H. Org. Lett. 2016, 18, 944–947. doi:10.1021/acs.orglett.5b03651 |
| 46. | Sasaki, T.; Moriyama, K.; Togo, H. J. Org. Chem. 2017, 82, 11727–11734. doi:10.1021/acs.joc.7b01433 |
| 25. | Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240–4243. doi:10.1021/ol501939m |
| 23. | Metternich, J. B.; Gilmour, R. J. Am. Chem. Soc. 2016, 138, 1040–1045. doi:10.1021/jacs.5b12081 |
| 24. | Yan, K.; Yang, D.; Wei, W.; Wang, F.; Shuai, Y.; Li, Q.; Wang, H. J. Org. Chem. 2015, 80, 1550–1556. doi:10.1021/jo502474z |
| 32. | Yu, Y.; Zhuang, S.; Liu, P.; Sun, P. J. Org. Chem. 2016, 81, 11489–11495. doi:10.1021/acs.joc.6b02155 |
| 33. | Ni, S.; Cao, J.; Mei, H.; Han, J.; Li, S.; Pan, Y. Green Chem. 2016, 18, 3935–3939. doi:10.1039/C6GC01027J |
| 30. | Wei, W.; Wen, J.; Yang, D.; Guo, M.; Wang, Y.; You, J.; Wang, H. Chem. Commun. 2015, 51, 768–771. doi:10.1039/C4CC08117J |
| 31. | Qiu, G.; Liu, T.; Ding, Q. Org. Chem. Front. 2016, 3, 510–515. doi:10.1039/C6QO00041J |
| 28. | Mi, X.; Wang, C.; Huang, M.; Wu, Y.; Wu, Y. J. Org. Chem. 2015, 80, 148–155. doi:10.1021/jo502220b |
| 29. | Yang, W.; Yang, S.; Li, P.; Wang, L. Chem. Commun. 2015, 51, 7520–7523. doi:10.1039/C5CC00878F |
| 26. | Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Org. Lett. 2014, 16, 3356–3359. doi:10.1021/ol5013839 |
| 27. | Fu, W.; Zhu, M.; Zou, G.; Xu, C.; Wang, Z.; Ji, B. J. Org. Chem. 2015, 80, 4766–4770. doi:10.1021/acs.joc.5b00305 |
© 2018 Sasaki et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)