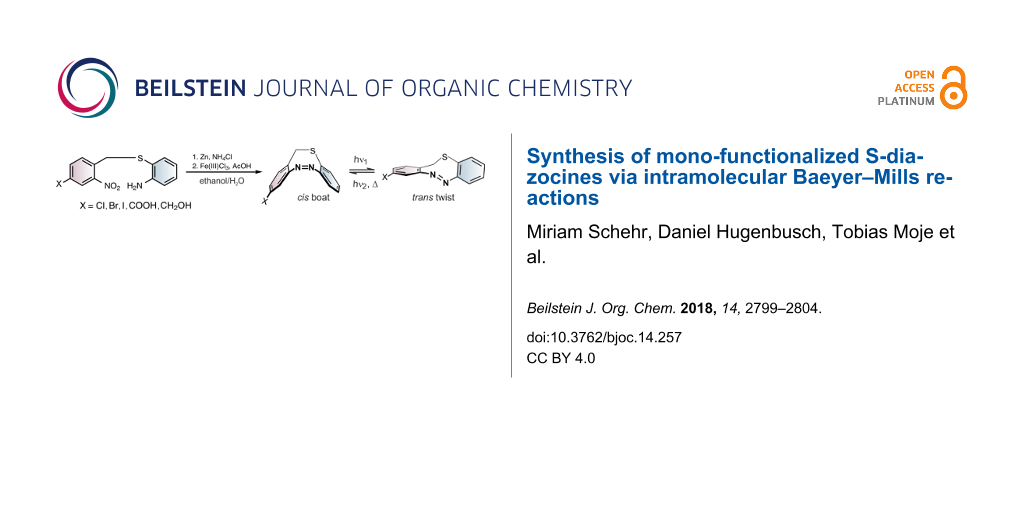
Abstract
Herein we report a reliable method to synthesize mono-functionalized S-diazocines in reproducible yields via intramolecular Baeyer–Mills reactions. Diazocines exhibit excellent photoswitchable properties. As opposed to azobenzenes they are more stable in their cis configuration. Particularly in photopharmacology mono-functionalized diazocines should be potentially useful and superior to the frequently used azobenzenes because the sterically more demanding cis configuration should be inactive, and the slender trans configuration should fit in a tight binding pocket of a receptor. Hence, it should be possible to administer the stabile inactive compound and switch it on at the site of illness with visible light. To date only a limited number of diazocine derivatives have been published of which most are symmetrically functionalized. Using the Baeyer–Mills reaction for the synthesis of diazocines opens a novel and convenient access to unsymmetrically substituted diazocines.

Graphical Abstract
Introduction
Photopharmacology is a promising, rapidly evolving field in medicine aiming at the development of drugs whose biological activity can be controlled with light [1-4]. There are several reported pharmacologically active compounds containing azobenzenes as photoswitchable units [5-7]. In most cases these drugs are active in the more stable trans configuration and therefore have to be switched to the inactive form before administration. Unfortunately, the cis form of azobenzenes is not stable. In most cases, the cis isomer thermally isomerizes back to the active trans within several hours. Therefore, spatiotemporal control of activation with light is difficult to achieve. On this account, bridged azobenzenes (diazocines), would be more suitable as photoswitches in pharmacologically active compounds [8]. Besides their superior photophysical properties, diazocines are more stable in the cis configuration and could therefore be administered in their inactive form and switched on at the site of interest. However, applications of diazocines have been rather limited due to low yields and difficult syntheses. Moreover, the preparation of unsymmetrically functionalized diazocines, which are essential precursors for incorporation in a drug, are especially challenging because most synthetic approaches involve homocoupling steps for the synthesis of the bridge as well as for the formation of the azo function [9-16]. In Table 1 previous synthesis approaches are compared to the synthesis approach in the present study.
Table 1: Comparison of previous diazocine syntheses approaches compared to the present study discussed in this publication.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-257-i2.png?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-257-i3.png?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-257-i4.png?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-257-i5.png?max-width=637&scale=1.0)
Recently, we reported the synthesis of a diazocine where the –CH2–CH2– bridge is replaced by a –CH2–S– bridge (S-diazocine) using an ultrasound-supported reductive azocoupling with lead [17]. In this reaction a small amount of S-diazocine is directly formed and a larger fraction of the azoxy compound is obtained, which can be reduced in a second synthetic step using lead in a ball mill. However, these reaction conditions are not suitable for functionalized S-diazocines due to poor yields and low reproducibility. Using halogenated precursors for the reductive azocoupling with lead a large amount of byproduct (details see Supporting Information File 1) is formed, which lowers the overall yield of S-diazocines 1–3 considerably. Further problems arise from the difficult separation of the azoxy compound from other byproducts. The yields of the azoxy reduction in the ball mill are varying between 18–46%. S-Diazocines functionalized with a carboxylic acid 4 or a benzyl alcohol 5 could not be obtained using the reductive azo coupling. For the benzyl alcohol 5 a variety of different, unidentified byproducts formed and in case of the carboxylic acid 4 only the starting material was retrieved. We now present a reliable one-pot-like synthesis of mono-functionalized S-diazocines in reproducible yields via intramolecular Baeyer–Mills reaction.
Results and Discussion
The Baeyer–Mills reaction is a quite common method, particularly to synthesize unsymmetrically substituted azobenzenes, albeit not widely used to prepare diazocine derivatives [18,19].
Towards the synthesis of S-diazocines (Figure 1), two general approaches could be derived from the well investigated azobenzene preparation methods: a) Reductive coupling of two nitro groups or b) oxidative coupling of two amino groups [20]. To cleanly form N=N bonds by reduction of dinitro compounds, strongly basic conditions have to be applied. Unfortunately, the benzylic position of the bridge is deprotonated under these conditions, leading to a plethora of side products. Formation of the N=N double bond by oxidation of diamines [21-23] cannot be applied either, because the sulfur bridge is oxidized to the sulfoxide or sulfone. Hence, neither reductive nor oxidative conditions are compatible with the –CH2-S– bridge. An alternative strategy, that applies weakly acidic conditions is the condensation of aryl nitroso compounds with anilines. This Baeyer–Mills reaction was discovered by Baeyer in 1874 [24] and further investigated by Mills [25] and Bamberger [26].
![[1860-5397-14-257-1]](/bjoc/content/figures/1860-5397-14-257-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cis–trans isomerization of mono-functionalized S-diazocines 1–5.
Figure 1: Cis–trans isomerization of mono-functionalized S-diazocines 1–5.
The S-diazocines 1–5 were obtained using a 2–3 step synthesis (Scheme 1). To synthesize the nitro and amino-substituted precursors 13–16, disulfide 12 was reduced with sodium borohydride and in situ reacted with benzyl bromides 8–11, which were commercially available or synthesized by radical bromination. The S-diazocines precursors 13–16 were then subjected to the intramolecular Baeyer–Mills reaction yielding the S-diazocines 1–4. In the first step the nitro group was reduced with zinc powder to the hydroxylamine and subsequently in situ oxidized to the nitroso compound using iron(III) chloride.
![[1860-5397-14-257-i1]](/bjoc/content/inline/1860-5397-14-257-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction conditions: i) MeCN, AIBN, NBS; ii) NaBH4, THF; #commercially available iii) BH3·THF complex, THF, *product 17 is not stable; iv) 1. Zinc powder, ammonium chloride, ethanol, 2. Fe(III)Cl3 hexahydrate, H2O/ethanol, acetic acid.
Scheme 1: Reaction conditions: i) MeCN, AIBN, NBS; ii) NaBH4, THF; #commercially available iii) BH3·THF compl...
After addition of acetic acid the reaction was allowed to proceed for several hours at room temperature to form the azo bond. For the benzyl alcohol-functionalized S-diazocine 5, the carboxylic acid of the precursor 16 was reduced using a borane tetrahydrofuran complex and immediately reacted in the Baeyer–Mills reaction without further purification. In Table 2 the yields using the Baeyer–Mills reaction are compared with the yields using the reductive azo condensation with lead [17].
Compared to the reductive azo condensation, the yields obtained using the Baeyer–Mills reaction are higher, more reproducible and applicable for an enlarged number of functional groups. Using this method, five different mono-functionalized S-diazocines 1–5 could be synthesized, which are suitable starting materials for further functionalization such as cross coupling, peptide coupling or further functionalization.
The photoswitchable properties of the S-diazocines 1–5 were investigated using UV–vis and NMR spectroscopic methods. The photostationary states (PSS), half-lives (t1/2) and absorption maxima (λmax) were recorded in acetone and are summarized in Table 3.
Table 3: Photostationary states, absorption maxima and half-lives of S-diazocines 1–5 determined with UV–vis and NMR spectroscopy in acetone.
| molecule | X | PSS (405 nm) | λmax (cis) | λmax (trans) | t1/2 (NMR) |
|---|---|---|---|---|---|
| 1 | Cl | 64% | 392 nm | 516 nm | 3.6 d |
| 2 | Br | 60% | 389 nm | 516 nm | 2.8 d |
| 3 | I | 51% | 393 nm | 515 nm | 3.3 d |
| 4 | COOH | 67% | 393 nm | 516 nm | 6.7 d |
| 5 | CH2OH | 65% | 393 nm | 508 nm | 24.8 h |
The photostationary states (PSS) upon irradiation with 405 nm are between 51% and 67% trans, and thus similar to the PSS of the parent system (70%) [17]. The absorption maxima of the nπ* excitation of the cis isomers are between 389 nm and 393 nm. For the trans isomers the nπ* absorption maxima are in a range between 508 nm to 516 nm (for UV–vis spectra measured in acetone see Supporting Information File 1). The half-lives vary from 24.8 h to 6.7 d.
For the carboxylic acid functionalized S-diazocine 4 the two isomers generated after irradiation with 405 nm could be separated by chromatography and the UV–vis spectra of the pure cis and trans isomers could be measured in an acetonitrile/water mixture. Figure 2 shows the UV spectra of the pure cis and trans isomers of 4.
![[1860-5397-14-257-2]](/bjoc/content/figures/1860-5397-14-257-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV spectra of the S-diazocine 4 in cis (black) and in trans (red) configuration after irradiation with 405 nm and separation by chromatography measured in a mixture of acetonitrile and water.
Figure 2: UV spectra of the S-diazocine 4 in cis (black) and in trans (red) configuration after irradiation w...
The nπ* band of the cis isomer displays an absorption maxima at λmax = 392 nm. Upon irradiation with 405 nm to the trans isomer the nπ* band is shifted bathochromically to λmax = 512 nm. The trans isomer exhibits also an additional absorption maximum in the same area as the cis isomer at 369 nm, which is the reason for the low cis-to-trans conversion when compared to the C-diazocine (>90% trans) [8]. The back isomerization is initiated quantitatively with light within the range of 500–660 nm. Hence, the trans-to-cis isomerization can be performed with red light which is important for medical applications, because blood-supported tissue is transparent in the far red region [27].
For the iodo-functionalized S-diazocine 3 single crystals could be obtained, which were characterized by single crystal X-ray diffraction. The structure of 3 is the first crystal structure of a S-diazocine reported in the Cambridge Structural Database (CSD; version 5.39; Feb. 2018) [28].
This compound crystallizes in the monoclinic space group P21/c with Z = 4 and all atoms in general positions (specification see Supporting Information File 1). The molecule exhibits a cis boat conformation with an dihedral angle of 72.24(8)° between both phenyl rings (Figure 3). Each of these rings is planar with maximum deviations from the mean plane calculated through the C atoms of 0.014(2) and 0.009(2) Å. The N=N bond lengths of the azo group of 1.251(4) Å corresponds to literature values and the CNNC torsion angle amounts to 3.7(4)°, which proves that this unit is nearly coplanar. In contrast, the CCSC fragment is twisted by 22.9(3)°. The structure of the title compound 3 can be compared with the structure of 5,6-dihydrobenzo[c,g][1,2]-diazocine (18) already reported in the literature [8]. In this compound the CNNC fragment is more planar with an torsion angle of 0.7(2)°. The dihedral angle between the phenyl rings of 85.00(6)° is larger compared to the S-diazocine 3. The ethylene group in the 8-membered ring is disordered in two orientations with torsion angles of 47.9(1) and 37.7(1)°, which means that the overall molecule is more distorted in the C-diazocine than in the sulfur analogue.
![[1860-5397-14-257-3]](/bjoc/content/figures/1860-5397-14-257-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Left: crystal structure of the iodo-functionalized S-diazocine 3. Right: crystal structure of the unfunctionalized C-diazocine 18 [8].
Figure 3: Left: crystal structure of the iodo-functionalized S-diazocine 3. Right: crystal structure of the u...
Conclusion
The Baeyer–Mills reaction as the key step in our approach provides an easy one-pot-like synthesis for mono-functionalized S-diazocines. Using this method, five different mono-functionalized S-diazocines 1–5 have been synthesized and were investigated regarding their photochemical properties. The S-diazocines 1–5 are potential precursors for the incorporation in photopharmacophores using cross-coupling, peptide coupling or further functionalization methods such as nucleophilic substitution reaction of the benzyl alcohol 5.
The yield determining steps prior to the Baeyer–Mills reaction are the formation of the hydroxylamine with zinc and the oxidation from the hydroxylamine to the nitroso compound. These reaction steps are difficult to monitor and furthermore the S-diazocine yield depends on the nature of substituents. However, the overall yields and the reliability of the formation of the azo group are superior to previously reported methods. All S-diazocines 1–5 are thermodynamically more stable in their cis configuration. Irradiation with violet light (405 nm) converts the cis into the trans isomer with 50–70% yield. Back-reaction with green to red light (500–660 nm) is quantitative. Thermal half-lives of the metastable trans isomer are between 1–7 days. These properties are ideal for applications in photopharmacology where the more bulky cis state is the inactive form and the trans isomer is the active configuration.
Supporting Information
CCDC-1857720 (3) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
| Supporting Information File 1: Analytical equipment, experimental procedures, spectral data and crystallographic data. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Mayer, G.; Heckel, A. Angew. Chem., Int. Ed. 2006, 45, 4900–4921. doi:10.1002/anie.200600387
Return to citation in text: [1] -
Beharry, A. A.; Woolley, G. A. Chem. Soc. Rev. 2011, 40, 4422–4437. doi:10.1039/c1cs15023e
Return to citation in text: [1] -
Szymański, W.; Beierle, J. M.; Kistemaker, H. A. V.; Velema, W. A.; Feringa, B. L. Chem. Rev. 2013, 113, 6114–6178. doi:10.1021/cr300179f
Return to citation in text: [1] -
Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. doi:10.1021/acs.chemrev.8b00037
Return to citation in text: [1] -
Broichhagen, J.; Frank, J. A.; Trauner, D. Acc. Chem. Res. 2015, 48, 1947–1960. doi:10.1021/acs.accounts.5b00129
Return to citation in text: [1] -
Velema, W. A.; Szymanski, W.; Feringa, B. L. J. Am. Chem. Soc. 2014, 136, 2178–2191. doi:10.1021/ja413063e
Return to citation in text: [1] -
Lerch, M. M.; Hansen, M. J.; van Dam, G. M.; Szymanski, W.; Feringa, B. L. Angew. Chem., Int. Ed. 2016, 55, 10978–10999. doi:10.1002/anie.201601931
Return to citation in text: [1] -
Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d
Return to citation in text: [1] [2] [3] [4] -
Duval, H. Bull. Soc. Chim. Fr. 1910, 7, 727.
Return to citation in text: [1] [2] -
Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007
Return to citation in text: [1] [2] -
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383
Return to citation in text: [1] [2] -
Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1
Return to citation in text: [1] [2] -
Tellkamp, T.; Shen, J.; Okamoto, Y.; Herges, R. Eur. J. Org. Chem. 2014, 5456–5461. doi:10.1002/ejoc.201402541
Return to citation in text: [1] [2] -
Deo, C.; Bogliotti, N.; Métivier, R.; Retailleau, P.; Xie, J. Chem. – Eur. J. 2016, 22, 9092–9096. doi:10.1002/chem.201601400
Return to citation in text: [1] [2] -
Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685
Return to citation in text: [1] [2] -
Meesook, J.; Joshi, D. K.; Yalagala, R. S.; Vanloon, J.; Simionescu, R.; Lough, A. J.; Gordon, H. L.; Yan, H. ChemistrySelect 2018, 3, 2697–2701. doi:10.1002/slct.201703126
Return to citation in text: [1] [2] -
Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846
Return to citation in text: [1] [2] [3] [4] -
Priewisch, B.; Rück-Braun, K. J. Org. Chem. 2005, 70, 2350–2352. doi:10.1021/jo048544x
Return to citation in text: [1] -
Gowenlock, B. G.; Richter-Addo, G. B. Chem. Rev. 2004, 104, 3315–3340. doi:10.1021/cr030450k
Return to citation in text: [1] -
Schündehütte, K. H. Diarylazoverbindungen. In Methoden der Organischen Chemie, Band X, Teil 3, Stickstoffverbindungen I; Müller, E., Ed.; Houben Weyl, 1965; pp 332–381.
Return to citation in text: [1] -
Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786
Return to citation in text: [1] -
Reuter, R.; Hostettler, N.; Neuburger, M.; Wegner, H. A. Eur. J. Org. Chem. 2009, 5647–5652. doi:10.1002/ejoc.200900861
Return to citation in text: [1] -
Zhu, Y.; Shi, Y. Org. Lett. 2013, 15, 1942–1945. doi:10.1021/ol4005917
Return to citation in text: [1] -
Baeyer, A. Ber. Dtsch. Chem. Ges. 1874, 7, 1638–1640. doi:10.1002/cber.187400702214
Return to citation in text: [1] -
Mills, C. J. Chem. Soc., Trans. 1895, 67, 925–933. doi:10.1039/CT8956700925
Return to citation in text: [1] -
Bamberger, E. Ber. Dtsch. Chem. Ges. 1896, 29, 102–104. doi:10.1002/cber.18960290119
Return to citation in text: [1] -
Stolik, S.; Delgado, J. A.; Pérez, A.; Anasagasti, L. J. Photochem. Photobiol., B 2000, 57, 90–93. doi:10.1016/S1011-1344(00)00082-8
Return to citation in text: [1] -
Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438
Return to citation in text: [1]
| 17. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 26. | Bamberger, E. Ber. Dtsch. Chem. Ges. 1896, 29, 102–104. doi:10.1002/cber.18960290119 |
| 17. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 1. | Mayer, G.; Heckel, A. Angew. Chem., Int. Ed. 2006, 45, 4900–4921. doi:10.1002/anie.200600387 |
| 2. | Beharry, A. A.; Woolley, G. A. Chem. Soc. Rev. 2011, 40, 4422–4437. doi:10.1039/c1cs15023e |
| 3. | Szymański, W.; Beierle, J. M.; Kistemaker, H. A. V.; Velema, W. A.; Feringa, B. L. Chem. Rev. 2013, 113, 6114–6178. doi:10.1021/cr300179f |
| 4. | Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. doi:10.1021/acs.chemrev.8b00037 |
| 24. | Baeyer, A. Ber. Dtsch. Chem. Ges. 1874, 7, 1638–1640. doi:10.1002/cber.187400702214 |
| 9. | Duval, H. Bull. Soc. Chim. Fr. 1910, 7, 727. |
| 10. | Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007 |
| 11. | Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383 |
| 12. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 13. | Tellkamp, T.; Shen, J.; Okamoto, Y.; Herges, R. Eur. J. Org. Chem. 2014, 5456–5461. doi:10.1002/ejoc.201402541 |
| 14. | Deo, C.; Bogliotti, N.; Métivier, R.; Retailleau, P.; Xie, J. Chem. – Eur. J. 2016, 22, 9092–9096. doi:10.1002/chem.201601400 |
| 15. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 16. | Meesook, J.; Joshi, D. K.; Yalagala, R. S.; Vanloon, J.; Simionescu, R.; Lough, A. J.; Gordon, H. L.; Yan, H. ChemistrySelect 2018, 3, 2697–2701. doi:10.1002/slct.201703126 |
| 8. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 20. | Schündehütte, K. H. Diarylazoverbindungen. In Methoden der Organischen Chemie, Band X, Teil 3, Stickstoffverbindungen I; Müller, E., Ed.; Houben Weyl, 1965; pp 332–381. |
| 8. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 5. | Broichhagen, J.; Frank, J. A.; Trauner, D. Acc. Chem. Res. 2015, 48, 1947–1960. doi:10.1021/acs.accounts.5b00129 |
| 6. | Velema, W. A.; Szymanski, W.; Feringa, B. L. J. Am. Chem. Soc. 2014, 136, 2178–2191. doi:10.1021/ja413063e |
| 7. | Lerch, M. M.; Hansen, M. J.; van Dam, G. M.; Szymanski, W.; Feringa, B. L. Angew. Chem., Int. Ed. 2016, 55, 10978–10999. doi:10.1002/anie.201601931 |
| 21. | Takeda, Y.; Okumura, S.; Minakata, S. Angew. Chem., Int. Ed. 2012, 51, 7804–7808. doi:10.1002/anie.201202786 |
| 22. | Reuter, R.; Hostettler, N.; Neuburger, M.; Wegner, H. A. Eur. J. Org. Chem. 2009, 5647–5652. doi:10.1002/ejoc.200900861 |
| 23. | Zhu, Y.; Shi, Y. Org. Lett. 2013, 15, 1942–1945. doi:10.1021/ol4005917 |
| 10. | Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007 |
| 16. | Meesook, J.; Joshi, D. K.; Yalagala, R. S.; Vanloon, J.; Simionescu, R.; Lough, A. J.; Gordon, H. L.; Yan, H. ChemistrySelect 2018, 3, 2697–2701. doi:10.1002/slct.201703126 |
| 17. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 28. | Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438 |
| 14. | Deo, C.; Bogliotti, N.; Métivier, R.; Retailleau, P.; Xie, J. Chem. – Eur. J. 2016, 22, 9092–9096. doi:10.1002/chem.201601400 |
| 18. | Priewisch, B.; Rück-Braun, K. J. Org. Chem. 2005, 70, 2350–2352. doi:10.1021/jo048544x |
| 19. | Gowenlock, B. G.; Richter-Addo, G. B. Chem. Rev. 2004, 104, 3315–3340. doi:10.1021/cr030450k |
| 8. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 11. | Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383 |
| 8. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 12. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 13. | Tellkamp, T.; Shen, J.; Okamoto, Y.; Herges, R. Eur. J. Org. Chem. 2014, 5456–5461. doi:10.1002/ejoc.201402541 |
| 15. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 17. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 27. | Stolik, S.; Delgado, J. A.; Pérez, A.; Anasagasti, L. J. Photochem. Photobiol., B 2000, 57, 90–93. doi:10.1016/S1011-1344(00)00082-8 |
© 2018 Schehr et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)