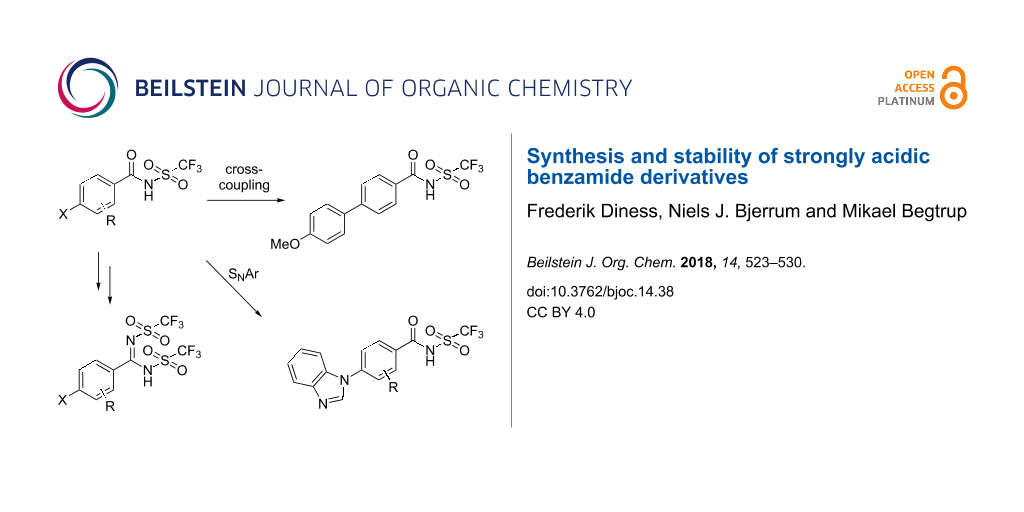
Abstract
Reactivity studies of strong organic acids based on the replacement of one or both of the oxygens in benzoic acids with the trifluoromethanesulfonamide group are reported. Novel derivatives of these types of acids were synthesized in good yields. The generated N-triflylbenzamides were further functionalized through cross-coupling and nucleophilic aromatic substitution reactions. All compounds were stable in dilute aqueous solutions. Studies of stability under acidic and basic conditions are also reported.

Graphical Abstract
Introduction
Very strong organic acids are interesting as catalysts for chemical reactions [1,2] and for facilitation of proton conduction [3]. In order to enable their incorporation into functional materials (e.g., polymers), these acids need additional functionality in the form of a reactive group, and the chemistry applied for functionalization must be compatible with their strong acidic nature. Many types of strong organic acids such as triflic acid (7) or trifluoroacetic acid (3) are not readily modified, and changing substituents on these acids will heavily impact their pKa value (Figure 1). In contrast, halogenated benzoic acid derivatives are easily functionalized through cross-coupling [4,5] or nucleophilic aromatic substitution reactions (SNAr) [6,7]. Benzoic acids (e.g., 2) are relatively weak acids, even with highly electron-withdrawing substituents on the aromatic core [8]. Very strong benzoic acid derivatives (e.g., 4 and 6) have been synthesized by replacing one or both of the oxygens of the carboxylate group with the trifluoromethanesulfonamide (1) group [9-11].
![[1860-5397-14-38-1]](/bjoc/content/figures/1860-5397-14-38-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Acid strength (pKa) of various organic acids in acetonitrile or water (nr = not reported) [12-14].
Figure 1: Acid strength (pKa) of various organic acids in acetonitrile or water (nr = not reported) [12-14].
Interestingly, these types of compounds have attracted little attention and have not thoroughly been explored with regards to their applications. Few publications report the application of N-triflylbenzamides as benzoic acid bioisosteres in receptor antagonists and enzyme inhibitors (Figure 2) [15-18]. The reported derivatives all displayed activity, but only with similar or reduced potency compared to the corresponding benzoic acid derivatives. Application of deprotonated N-triflylbenzamide derivatives as counter anions in supramolecular crown ether compounds for metal ion extraction has also been reported by one group (Figure 2) [19-25]. All the N-triflylbenzamide constructs generated for this purpose have proved superior to the corresponding benzoate derivatives with regards to metal ion extraction capability.
![[1860-5397-14-38-2]](/bjoc/content/figures/1860-5397-14-38-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Examples of functional molecules containing an N-triflylbenzamide.
Figure 2: Examples of functional molecules containing an N-triflylbenzamide.
The N-triflylbenzamides are simple to generate by the reaction between trifluoromethanesulfonamide (1) and an activated benzoyl derivative such as benzoyl chlorides (e.g., 8a, Scheme 1) [9,15]. The reactions are generally high yielding and the products are simple to isolate in their protonated form by recrystallization. The generation of N-triflylbenzamides by direct reaction of various triflyl derivatives with benzamides or benzoic acids has been less systematically explored. Most compounds synthesized by these approaches are N-alkylated or N-arylated and have been formed unintentionally as byproducts [26-33]. Finally, two examples of syntheses via palladium-catalyzed carbonylation of trifluoromethanesulfonamide (1) have been reported [34,35]. The N-triflylbenzamides have only been explored as substrates in a few reactions. A recent report describes using N-triflylbenzamide as a directing group in a ruthenium-catalyzed C–H activation reaction [36]. Most other reported transformations pass via the imidoyl chloride intermediates (equivalent to 10) to the amidine derivatives, which have been used in rearrangement reaction studies [9,37-39]. By these means also N,N’-bis(triflyl)benzimidamides (also termed benzamidines) have been generated, but only one report describes their syntheses [10]. The N-triflylbenzamides are stronger acids than any of the carboxylic acids, including trifluoroacetic acid (3). The N,N’-bis(triflyl)benzimidamides are very strong organic acids, much stronger than p-toluenesulfonic acid (5) which is commonly used as a soluble organic acid catalyst in chemical reactions. Remarkably, neither the N-triflylbenzamides nor the N,N’-bis(triflyl)benzimidamides have been studied as Brønsted acid catalysts. Their chemical stability including compatibility with conditions applied to common chemical transformations has not been described. With the prospect of using these strong benzoic acid derivatives for enhancing proton conductivity in proton-exchange membrane (PEM) fuel cells [3,40] we have examined their compatibility with chemical transformations as well as their stability towards hydrolytic conditions.
Results and Discussion
In these studies, four substituted N-triflylbenzamides 9a–d were synthesized by the reaction of the corresponding benzoyl chloride with trifluoromethanesulfonamide (1, Scheme 1). The known 4-fluoro-N-triflylbenzamide (9a) was synthesized according to the previously reported method [9] and with few adjustments this methodology was also applied for the generation of three new derivatives 9b–d, which were obtained in good yields. The 4-bromo derivative 9d was further converted into the N,N’-bis(triflyl)benzimidamide 12 by formation of the corresponding imidoyl chlorides 10 with PCl5 in POCl3, followed by the additional reaction with trifluoromethanesulfonamide (1) and protonation by sulfuric acid (Scheme 1) [10].
![[1860-5397-14-38-i1]](/bjoc/content/inline/1860-5397-14-38-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the strongly acidic benzamide derivatives.
Scheme 1: Synthesis of the strongly acidic benzamide derivatives.
In recent years, we have reported high yielding catalyst-free N-arylation by SNAr reaction of mono- or perfluorobenzene derivatives [41-43]. Hence, it was proposed that the 4-fluoro and the pentafluorobenzamide derivatives 9a and 9c could be functionalized through SNAr reactions. Thus, compounds 9a and 9c were reacted with benzimidazole under previously developed conditions (Scheme 2) [41,42]. The benzimidazole was chosen as model nucleophile for polybenzimidazole, a polymer commonly applied as a membrane in PEM fuel cells [40]. These reactions provided the 4-benzimidazolyl derivatives 13 and 14 in good yields and the N-triflylbenzamide group proved to be stable under these reaction conditions. Gratifyingly it was possible to perform selective mono-substitution of the pentafluoro derivative regioselectively in the 4-position, affording only compound 14. This is in line with previous observations of SNAr reactions on pentafluorobenzene derivatives [42]. Due to the zwitterionic nature of the products, reversed-phase chromatography was chosen to simplify the purifications.
![[1860-5397-14-38-i2]](/bjoc/content/inline/1860-5397-14-38-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: SNAr reactions of fluoro-substituted benzamide derivatives.
Scheme 2: SNAr reactions of fluoro-substituted benzamide derivatives.
The N-triflylbenzamide group also proved stable to cross-coupling reaction conditions, as exemplified by a palladium-catalyzed Suzuki–Miyaura reaction of the 4-bromo-substituted derivative 9d (Scheme 3). The corresponding N,N’-bis(triflyl)benzimidamide derivative 12 was also tested under identical conditions. The reaction proceeded with high conversion, but surprisingly the major product of this reaction was also the N-triflylbenzamide 15. Additional experiments revealed that the N,N’-bis(triflyl)benzimidamide group was unstable in aqueous basic conditions.
![[1860-5397-14-38-i3]](/bjoc/content/inline/1860-5397-14-38-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cross-coupling reactions of N-triflylbenzoic acid derivatives.
Scheme 3: Cross-coupling reactions of N-triflylbenzoic acid derivatives.
This led to concerns about the stability of the products in general. In addition, during characterization by NMR it was also noted that some samples of the N-triflylbenzamide products contained small amounts of the parent benzoic acid, despite prior purification by recrystallization. Further studies revealed that the content of benzoic acid increased over time in the solutions and the conversion rate was concentration dependent, the reason being simple hydrolysis auto-catalyzed by the acids themselves. This phenomenon was also observed for N,N’-bis(triflyl)benzimidamide 12 but at slower rate. Hence, an elaborated study of the products’ stability towards acid or base promoted hydrolysis was undertaken (Scheme 4). Dilute aqueous solutions (0.5 mg/mL) of the N-triflylbenzamides displayed no sign of degeneration even after 24 h. The compounds also remained fully intact in 0.5 M aqueous NaOH. In solutions of 0.5 M aqueous HCl the compounds very slowly degraded over weeks. The N,N’-bis(triflyl)benzimidamide 12 was also stable in dilute aqueous solutions and was even more stable in 0.5 M aqueous HCl than the corresponding N-triflylbenzamide 9d. Hence, in the acid-catalyzed hydrolysis reaction of 12 the 4-bromo-N-triflylbenzamide (9d) was only detected as a trace as 9d converted faster to 4-bromobenzoic acid than compound 12 converted to 9d. In contrast, 0.5 M aqueous NaOH rapidly hydrolyzed the N,N’-bis(triflyl)benzimidamide 12 to yield the base-stable N-triflylbenzamide 9d.
![[1860-5397-14-38-i4]](/bjoc/content/inline/1860-5397-14-38-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Hydrolysis rates of the 4-bromobenzoic acid derivatives.
Scheme 4: Hydrolysis rates of the 4-bromobenzoic acid derivatives.
Neither the mono-substituted N-triflylbenzamides 9a and 9b nor the 4-bromo-N,N’-bis(triflyl)benzimidamide (12) were stable in heated methanolic phosphoric acid, which is commonly used for operating PEM fuel cells (Figure 3) [44]. However, most surprisingly, the pentafluoro N-triflylbenzamide (9c) was found to be much more stable under these conditions, which is promising for future applications in proton conducting materials.
![[1860-5397-14-38-3]](/bjoc/content/figures/1860-5397-14-38-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Content (percent) of super acids (0.5 mg/mL) over time (hours) in H3PO4/H2O/MeOH 17:3:20 at 50 °C.
Figure 3: Content (percent) of super acids (0.5 mg/mL) over time (hours) in H3PO4/H2O/MeOH 17:3:20 at 50 °C.
Conclusion
In summary, it has been demonstrated that novel mono- or bis-trifluoromethanesulfonamide derivatives of benzoic acids bearing a reactive group (bromine or fluorine) may be generated in good yields and further functionalized through nucleophilic aromatic substitution or cross-coupling reactions. It was found that the products were stable in dilute aqueous solutions, but they slowly hydrolyzed in concentrated solutions or in the presence of other strong acids. The N-triflylbenzamides 9a–d were stable in basic aqueous solutions whereas the tested bromo-N,N’-bis(triflyl)benzimidamide 12 rapidly hydrolyzed to the corresponding bromo-N-triflylbenzamide 9d. Under conditions simulating ambient PEM fuel cells operation the 4-substituted benzamide derivatives 9a,b,d and 12 were about 50% degraded within 24 h, whereas the pentafluoro N-triflylbenzamide (9c) under the same conditions was less than 5% degraded.
Experimental
General aspects
All purchased chemicals were used without further purification. All solvents were HPLC grade. NMR data were recorded on a Bruker 500 MHz spectrometer at 298 K with methanol-d4, DMSO-d6 or hexafluorobenzene as internal standard. High-resolution mass spectrometry (HRMS) was performed on a Bruker MALDI–TOF spectrometer.
General procedure
Benzoyl chloride (10.0 mmol) in Et2O (10 mL) was added dropwise to a solution of trifluoromethanesulfonamide (1.5 g, 10.0 mmol) and triethylamine (3.5 mL, 25 mmol) in Et2O (30 mL) at 0 °C over 10 min. The mixture was stirred for 30 min and then heated to reflux for 2 h. The reaction was filtered and the solvent was removed from the filtrate. The crude product was mixed with 50% H2SO4 (aq, 50 mL) and the mixture stirred for 30 min. The precipitate was isolated and dried overnight. The product was purified by recrystallization from toluene.
4-Fluoro-N-((trifluoromethyl)sulfonyl)benzamide (9a) [9]: Synthesized by general procedure. The product was obtained as small clear crystalline flakes (1.88 g, 69%). mp 152–154 °C; 1H NMR (500 MHz, methanol-d4) δ 7.99 (dd, J = 9.0, 5.2 Hz, 2H), 7.29 (t, J = 8.8 Hz, 2H); 13C NMR (126 MHz, methanol-d4) δ 167.6 (d, J = 254.3 Hz), 166.0, 132.8 (d, J = 9.6 Hz), 129.1 (d, J = 3.1 Hz), 121.0 (q, J = 321.5 Hz), 117.1 (d, J = 22.7 Hz); 19F NMR (470 MHz, methanol-d4) δ −76.93 (s, 3F), −105.75 (s, 1F); HRMS (TOF) m/z: [M − H]− calcd for C8H4F4NO3S−, 269.9854; found, 269.9888.
4-Trifluoromethyl-N-((trifluoromethyl)sulfonyl)benzamide (9b): Synthesized by general procedure. The product was obtained as white powder (2.83 g, 88%). mp 174–179 °C; 1H NMR (500 MHz, methanol-d4) δ 8.09 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.1 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ 166.3, 136.5, 136.0 (q, J = 32.6 Hz), 130.6, 127.0 (q, J = 4.0 Hz), 125.0 (q, J = 271.9 Hz), 121.0 (q, J = 321.5 Hz); 19F NMR (470 MHz, methanol-d4) δ −64.31 (s, 3F), −77.06 (s, 3F); HRMS (TOF) m/z: [M − H]− calcd for C9H4F6NO3S−, 319.9822; found, 319.9869.
Pentafluoro-N-((trifluoromethyl)sulfonyl)benzamide (9c): Synthesized by general procedure. The product was obtained as clear needle-shaped crystal (2.40 g, 70%). mp 129–131°C; 13C NMR (126 MHz, methanol-d4) δ 158.6, 145.4 (dm, J = 257.0 Hz), 144.5 (dm, J = 256.8 Hz), 139.1 (dm, J = 250.0 Hz), 121.0 (q, J = 321.4 Hz), 111.9 (t, J = 17.6 Hz); 19F NMR (470 MHz, methanol-d4) δ −78.15 (s, 3F), −142.78 (d, J = 17.9 Hz, 2F), −152.25 (t, J = 19.2 Hz, 1F), −162.65 (t, J = 18.3 Hz, 2F); HRMS (TOF) m/z: [M − H]− calcd for C8F8NO3S−, 341.9477; found, 341.9520.
4-Bromo-N-((trifluoromethyl)sulfonyl)benzamide (9d): Synthesized by general procedure. The product was obtained as white powder (2.98 g, 90%). mp 156–158 °C; 1H NMR (500 MHz, methanol-d4) δ 7.83 (d, J = 8.8 Hz, 2H), 7.73 (d, J = 8.7 Hz, 2H); 13C NMR (126 MHz, methanol-d4) δ 166.5, 133.3, 131.9, 131. 6, 129.9, 121.0 (q, J = 321.5 Hz); 19F NMR (470 MHz, MeOD) δ −77.00 (s); HRMS (TOF) m/z: [M − H]− calcd for C8H479BrF3NO3S−, 329.9053; found, 329.9095.
Triethylamine salt of 4-bromo-N,N'-bis((trifluoromethyl)sulfonyl)benzimidamide (11): 4-Bromo-N-((trifluoromethyl)sulfonyl)benzamide (9d, 1.68 g, 5.0 mmol) was mixed with POCl3 (3 mL) and PCl5 (1.11 g). The reaction mixture was stirred at rt for 2 h and then at 50 °C for 30 min. The solvent was removed and the imidoyl chloride was recrystallized from heptane (1.71 g, 98%). The imidoyl chloride (1.06 g) was dissolved in dry acetonitrile (4 mL) and the solution was added dropwise to a mixture of trifluoromethanesulfonamide (450 mg) and triethylamine (0.85 mL) in dry acetonitrile (8 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min and then at rt for 5 h. The solvent was removed in vacuo and the residue was mixed with CH2Cl2 (30 mL) and 10% HCl (aq, 50 mL). The organic phase was separated and the water phase was extracted with CH2Cl2 (30 mL). The combined organic phase was washed with saturated NaHCO3 (aq) and dried with MgSO4. The solvent was removed in vacuo and the product was isolated as white powder (1.58 g, 93%). 1H NMR (500 MHz, methanol-d4) δ 7.73 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 3.20 (q, J = 7.3 Hz, 6H), 1.30 (t, J = 7.3 Hz, 9H); 13C NMR (126 MHz, methanol-d4) δ 170.0, 137.8, 132.3, 132.0, 127.6, 121.1 (q, J = 319.2 Hz), 48.0, 9.3; 19F NMR (470 MHz, MeOD) δ −80.67 (s); HRMS (TOF) m/z: [M − H]− calcd for C9H479BrF6N2O4S2−, 460.8706; found, 460.8769.
4-Bromo-N,N'-bis((trifluoromethyl)sulfonyl)benzimidamide (12): The trimethylamine salt of 4-bromo-N,N'-bis((trifluoromethyl)sulfonyl)benzimidamide (11, 0.50 g, 0.89 mmol) was mixed with H2SO4 (96%, 1.0 mL). The mixture was shaken for 30 min and extracted with CH2Cl2 (5 × 6 mL). The extract was dried with MgSO4 and the solvent removed in vacuo. The crude product was recrystallized from toluene and heptane to yield 12 as a white crystalline powder (367 mg, 0.79 mmol, 89%). 1H NMR (500 MHz, methanol-d4) δ 7.72 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 8.6 Hz, 2H); 13C NMR (126 MHz, methanol-d4) δ 170.1, 137.6, 132.23, 131.9, 127.6, 121.02 (q, J = 319.3 Hz); 19F NMR (470 MHz, MeOD) δ −80.60 (s); HRMS (TOF) m/z: [M − H]− calcd for C9H479BrF6N2O4S2−, 460.8706; found. 460.8766.
4-(1H-Benzo[d]imidazol-1-yl)-N-((trifluoromethyl)sulfonyl)benzamide (13): Cesium carbonate (163 mg, 5 equiv) was added to a glass vial containing benzimidazole (17.7 mg, 1.5 equiv) and 4-fluoro-N-((trifluoromethyl)sulfonyl)benzamide (9a, 27.1 mg, 0.1 mmol) in dry dimethylacetamide (1.0 mL). The reaction was heated to 120 °C for 12 h. The reaction was quenched with 1.0 M HCl (aq, 100 μL) and the solvent removed in vacuo. The crude product was dissolved in acetonitrile/water 1:1 and filtered through a Teflon syringe filter. The solution was transferred to a C18 gel column and purified by vacuum liquid chromatography (VLC, 0 to 90% CH3CN in 0.01 M HCl). After lyophilization the product was obtained as a white powder (17 mg, 46%). 1H NMR (500 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.20 (d, J = 8.5 Hz, 2H), 7.99–7.90 (m, 1H), 7.81 (d, J = 8.5 Hz, 3H), 7.64–7.53 (m, 2H), 4.53 (s, 2H); 13C NMR (126 MHz, DMSO-d6) δ 168.5, 142.3, 138.4, 136.0, 134.8, 131.4, 130.3, 126.0, 125.6, 124.0, 120.3 (q, J = 324.8 Hz), 116.6, 112.6; 19F NMR (470 MHz, DMSO-d6) δ −79.75 (s); HRMS (TOF) m/z: [M − H]− calcd for C15H9F3N3O3S−, 368.0322; found, 368.0367.
4-(1H-Benzo[d]imidazol-1-yl)-2,3,5,6-tetrafluoro-N-((trifluoromethyl)sulfonyl)benzamide (14): Sodium tert-butoxide (21 mg, 2.2 equiv) was added to a glass vial containing benzimidazole (11.8 mg, 0.1 mmol) in dry dimethylacetamide (1.0 mL). The mixture was stirred at rt for 1 min. The mixture was cooled to 0 °C and added to a solution of 9c (37.7 mg, 0.11 mmol) in dry dimethylacetamide (1.0 mL) under stirring at 0 °C. The reaction was allowed to reach rt and stirred for 1 h. The reaction was quenched with 1.0 M HCl (aq, 20 μL) and the solvent removed in vacuo. The crude product was dissolved in acetonitrile/water 1:1 and filtered through a Teflon syringe filter. The solution was transferred to a C18 gel column and purified by vacuum liquid chromatography (VLC, 0 to 90% CH3CN in 0.01 M HCl). After lyophilization the product was obtained as a white powder (30 mg, 81%). 1H NMR (500 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.83 (dt, J = 7.3, 3.6 Hz, 1H), 7.66–7.57 (m, 1H), 7.47–7.34 (m, 2H); 13C NMR (126 MHz, DMSO) δ 161.34, 143.9, 143.6 (dddd, J = 246.1, 13.0, 8.4, 4.1 Hz), 142.2 (ddm, J = 252.7, 16.4 Hz), 141.3, 137.6 (dm, J = 254.1 Hz), 133.1, 124.5, 123.6, 121.4 (t, J = 21.9 Hz), 120.0 (q, J = 323.7 Hz), 119. 5, 114.1 (t, J = 14.6 Hz), 111.3; 19F NMR (470 MHz, DMSO-d6) δ −80.53 (s), −145.56 (dd, J = 25.2, 12.3 Hz), −148.81 (dd, J = 24.1, 11.3 Hz); HRMS (TOF) m/z: [M − H]− calcd for C15H5F7N3O3S−, 439.9945; found, 434.0006.
4'-Methoxy-N-((trifluoromethyl)sulfonyl)-[1,1'-biphenyl]-4-carboxamide (15): Palladium(II) acetate (6 mg, 27 μmol), triphenylphosphine (20 mg, 76 μmol) and H2O (0.5 μL, 28 μmol) were mixed in degassed dimethylacetamide (1.0 mL). The catalyst was preformed by heating the mixture in a closed screw cap vial to 100 °C for 1 min (color changed from yellow to deep red). A part of the catalyst mixture (0.2 mL) was added to a microwave glass vial containing a degassed mixture of 9d (33.1 mg, 0.1 mmol), 4-methoxyphenylboronic acid (22.8 mg, 0.15 mmol, 1.5 equiv) and cesium carbonate (100 mg, 3 equiv) in dimethylacetamide (0.8 mL). The vial was closed with a teflon cap and heated to 80 °C for 12 h under stirring. The reaction was quenched with 1.0 M HCl (aq, 100 μL) and the solvent removed in vacuo. The crude product was dissolved in acetonitrile/water 1:1 and filtered through a Teflon syringe filter. The solution was transferred to a C18 gel column and purified by vacuum liquid chromatography (VLC, 0 to 90% CH3CN in 0.01 M HCl). After lyophilization the product was obtained as a white powder (11 mg, 31%). 1H NMR (500 MHz, DMSO-d6) δ 7.96 (d, J = 8.3 Hz, 2H), 7.69–7.59 (m, 4H), 7.03 (d, J = 8.8 Hz, 2H), 3.80 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.5, 159.2, 142.0, 135.7, 131.8, 129.2, 127.9, 125.4, 120.4 (q, J = 325.3 Hz). 114.4, 55.2; 19F NMR (470 MHz, DMSO-d6) δ −79.57 (s); HRMS (TOF) m/z: [M − H]− calcd for C15H5F7N3O3S−, 358.0366; found, 358.0408.
Supporting Information
| Supporting Information File 1: NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Akiyama, T. Chem. Rev. 2007, 107, 5744–5758. doi:10.1021/cr068374j
Return to citation in text: [1] -
Akiyama, T.; Mori, K. Chem. Rev. 2015, 115, 9277–9306. doi:10.1021/acs.chemrev.5b00041
Return to citation in text: [1] -
Peighambardoust, S. J.; Rowshanzamir, S.; Amjadi, M. Int. J. Hydrogen Energy 2010, 35, 9349–9384. doi:10.1016/j.ijhydene.2010.05.017
Return to citation in text: [1] [2] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Suzuki, A. J. Organomet. Chem. 1999, 576, 147–168. doi:10.1016/S0022-328X(98)01055-9
Return to citation in text: [1] -
Dehe, D.; Munstein, I.; Reis, A.; Thiel, W. R. J. Org. Chem. 2011, 76, 1151–1154. doi:10.1021/jo102063s
Return to citation in text: [1] -
Keipour, H.; Hosseini, A.; Afsari, A.; Oladee, R.; Khalilzadeh, M. A.; Ollevier, T. Can. J. Chem. 2016, 94, 95–104. doi:10.1139/cjc-2015-0300
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6
Return to citation in text: [1] [2] [3] [4] [5] -
Yagupolskii, L. M.; Petrik, V. N.; Kondratenko, N. V.; Sooväli, L.; Kaljurand, I.; Leito, I.; Koppel, I. A. J. Chem. Soc., Perkin Trans. 2 2002, 1950–1955. doi:10.1039/B204172C
Return to citation in text: [1] [2] [3] -
Koppel, I. A.; Burk, P.; Koppel, I.; Leito, I. J. Am. Chem. Soc. 2002, 124, 5594–5600. doi:10.1021/ja0255958
Return to citation in text: [1] -
Leito, I.; Kaljurand, I.; Koppel, I. A.; Yagupolskii, L. M.; Vlasov, V. M. J. Org. Chem. 1998, 63, 7868–7874. doi:10.1021/jo981124l
Return to citation in text: [1] -
Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; Pihl, V.; Koppel, I. A.; Leito, I. J. Phys. Org. Chem. 2013, 26, 162–170. doi:10.1002/poc.2946
Return to citation in text: [1] -
Trummal, A.; Lipping, L.; Kaljurand, I.; Koppel, I. A.; Leito, I. J. Phys. Chem. A 2016, 120, 3663–3669. doi:10.1021/acs.jpca.6b02253
Return to citation in text: [1] -
Lowe, J. A., III; Drozda, S. E.; McLean, S.; Bryce, D. K.; Crawford, R. T.; Zorn, S.; Morrone, J.; Appleton, T. A.; Lombardo, F. Bioorg. Med. Chem. Lett. 1995, 5, 1933–1936. doi:10.1016/0960-894X(95)00327-P
Return to citation in text: [1] [2] -
Hamada, Y.; Abdel-Rahman, H.; Yamani, A.; Nguyen, J.-T.; Stochaj, M.; Hidaka, K.; Kimura, T.; Hayashi, Y.; Saito, K.; Ishiurac, S.; Kiso, Y. Bioorg. Med. Chem. Lett. 2008, 18, 1649–1653. doi:10.1016/j.bmcl.2008.01.058
Return to citation in text: [1] -
Cervi, G.; Magnaghi, P.; Asa, D.; Avanzi, N.; Badari, A.; Borghi, D.; Caruso, M.; Cirla, A.; Cozzi, L.; Felder, E.; Galvani, A.; Gasparri, F.; Lomolino, A.; Magnuson, S.; Malgesini, B.; Motto, I.; Pasi, M.; Rizzi, S.; Salom, B.; Sorrentino, G.; Troiani, S.; Valsasina, B.; O’Brien, T.; Isacchi, A.; Donati, D.; D’Alessio, R. J. Med. Chem. 2014, 57, 10443–10454. doi:10.1021/jm501313x
Return to citation in text: [1] -
Althaus, J.; Hake, T.; Hanekamp, W.; Lehr, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 131–140. doi:10.1080/14756366.2016.1178246
Return to citation in text: [1] -
Talanov, V. S.; Talanova, G. G.; Bartsch, R. A. Tetrahedron Lett. 2000, 41, 8221–8224. doi:10.1016/S0040-4039(00)01457-X
Return to citation in text: [1] -
Talanov, V. S.; Talanova, G. G.; Gorbunova, M. G.; Bartsch, R. A. J. Chem. Soc., Perkin Trans. 2 2002, 209–215. doi:10.1039/b109638a
Return to citation in text: [1] -
Talanov, V. S.; Talanova, G. G.; Gorbunova, M. G.; Bartsch, R. A. Tetrahedron Lett. 2002, 43, 1629–1631. doi:10.1016/S0040-4039(02)00096-5
Return to citation in text: [1] -
Surowiec, M.; Custelcean, R.; Surowiec, K.; Bartsch, R. A. Tetrahedron 2009, 65, 7777–7783. doi:10.1016/j.tet.2009.07.006
Return to citation in text: [1] -
Yang, Y.; Arora, G.; Fernandez, F. A.; Crawford, J. D.; Surowiec, K.; Lee, E. K.; Bartsch, R. A. Tetrahedron 2011, 67, 1389–1397. doi:10.1016/j.tet.2010.12.006
Return to citation in text: [1] -
Boston, A. L.; Lee, E. K.; Surowiec, K.; Gega, J.; Bartsch, R. A. Tetrahedron 2012, 68, 8789–8794. doi:10.1016/j.tet.2012.07.103
Return to citation in text: [1] -
Zhou, H.; Connery, K. E.; Bartsch, R. A.; Moyer, B. A.; Haverlock, T. J.; Delmau, L. H. Solvent Extr. Ion Exch. 2013, 31, 683–696. doi:10.1080/07366299.2013.806754
Return to citation in text: [1] -
Hendrickson, J. B.; Bergeron, R.; Giga, A.; Sternbach, D. J. Am. Chem. Soc. 1973, 95, 3412–3413. doi:10.1021/ja00791a072
Return to citation in text: [1] -
Hendrickson, J. B.; Bergeron, R. Tetrahedron Lett. 1973, 14, 4607–4610. doi:10.1016/S0040-4039(01)87289-0
Return to citation in text: [1] -
Benati, L.; Nanni, D.; Spagnolo, P. J. Org. Chem. 1999, 64, 5132–5138. doi:10.1021/jo9901541
Return to citation in text: [1] -
Pampín, M. C.; Estévez, J. C.; Estévez, R. J.; Maestro, M.; Castedo, L. Tetrahedron 2003, 59, 7231–7243. doi:10.1016/S0040-4020(03)01073-1
Return to citation in text: [1] -
Shainyan, B. A.; Tolstikova, L. L.; Bel'skikh, A. V. Russ. J. Org. Chem. 2008, 44, 1121–1125. doi:10.1134/S1070428008080022
Return to citation in text: [1] -
Moskalik, M. Y.; Meshcheryakov, V. I.; Shainyan, B. A. Russ. J. Org. Chem. 2009, 45, 1644–1650. doi:10.1134/S1070428009110116
Return to citation in text: [1] -
Cody, J. A.; Ahmed, I.; Tusch, D. J. Tetrahedron Lett. 2010, 51, 5585–5587. doi:10.1016/j.tetlet.2010.08.058
Return to citation in text: [1] -
Wu, C.; Li, P.; Shi, X.; Pan, X.; Wu, J. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o382. doi:10.1107/S1600536811001085
Return to citation in text: [1] -
Wu, X.; Rönn, R.; Gossas, T.; Larhed, M. J. Org. Chem. 2005, 70, 3094–3098. doi:10.1021/jo050080v
Return to citation in text: [1] -
Urriolabeitia, E. P.; Laga, E.; Cativiela, C. Beilstein J. Org. Chem. 2012, 8, 1569–1575. doi:10.3762/bjoc.8.179
Return to citation in text: [1] -
Petrova, E.; Rasina, D.; Jirgensons, A. Eur. J. Org. Chem. 2017, 1773–1779. doi:10.1002/ejoc.201601582
Return to citation in text: [1] -
Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Sokolenko, L. V.; Chernega, A. N.; Rusanov, E. B.; Tsymbal, I. F. J. Fluorine Chem. 2007, 128, 515–523. doi:10.1016/j.jfluchem.2007.01.002
Return to citation in text: [1] -
Yagupolskii, L. M.; Maletina, I. I.; Sokolenko, L. V.; Vlasenko, Y. G.; Buth, S. A. J. Fluorine Chem. 2008, 129, 486–492. doi:10.1016/j.jfluchem.2008.03.001
Return to citation in text: [1] -
Yagupolskii, L. M.; Maletina, I. I.; Sokolenko, L. V.; Vlasenko, Y. G.; Drozdova, M. V.; Polovinko, V. V. J. Fluorine Chem. 2010, 131, 238–247. doi:10.1016/j.jfluchem.2009.10.019
Return to citation in text: [1] -
Asensio, J. A.; Sánchez, E. M.; Gómez-Romero, P. Chem. Soc. Rev. 2010, 39, 3210–3239. doi:10.1039/b922650h
Return to citation in text: [1] [2] -
Diness, F.; Fairlie, D. P. Angew. Chem., Int. Ed. 2012, 51, 8012–8016. doi:10.1002/anie.201202149
Return to citation in text: [1] [2] -
Diness, F.; Begtrup, M. Org. Lett. 2014, 16, 3130–3133. doi:10.1021/ol5012554
Return to citation in text: [1] [2] [3] -
Jacobsen, C. B.; Meldal, M.; Diness, F. Chem. – Eur. J. 2017, 23, 846–851. doi:10.1002/chem.201604098
Return to citation in text: [1] -
Wang, J.-T.; Wainright, J. S.; Savinell, R. F.; Litt, M. J. Appl. Electrochem. 1996, 26, 751–756. doi:10.1007/bf00241516
Return to citation in text: [1]
| 41. | Diness, F.; Fairlie, D. P. Angew. Chem., Int. Ed. 2012, 51, 8012–8016. doi:10.1002/anie.201202149 |
| 42. | Diness, F.; Begtrup, M. Org. Lett. 2014, 16, 3130–3133. doi:10.1021/ol5012554 |
| 43. | Jacobsen, C. B.; Meldal, M.; Diness, F. Chem. – Eur. J. 2017, 23, 846–851. doi:10.1002/chem.201604098 |
| 9. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6 |
| 10. | Yagupolskii, L. M.; Petrik, V. N.; Kondratenko, N. V.; Sooväli, L.; Kaljurand, I.; Leito, I.; Koppel, I. A. J. Chem. Soc., Perkin Trans. 2 2002, 1950–1955. doi:10.1039/B204172C |
| 1. | Akiyama, T. Chem. Rev. 2007, 107, 5744–5758. doi:10.1021/cr068374j |
| 2. | Akiyama, T.; Mori, K. Chem. Rev. 2015, 115, 9277–9306. doi:10.1021/acs.chemrev.5b00041 |
| 8. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 10. | Yagupolskii, L. M.; Petrik, V. N.; Kondratenko, N. V.; Sooväli, L.; Kaljurand, I.; Leito, I.; Koppel, I. A. J. Chem. Soc., Perkin Trans. 2 2002, 1950–1955. doi:10.1039/B204172C |
| 6. | Dehe, D.; Munstein, I.; Reis, A.; Thiel, W. R. J. Org. Chem. 2011, 76, 1151–1154. doi:10.1021/jo102063s |
| 7. | Keipour, H.; Hosseini, A.; Afsari, A.; Oladee, R.; Khalilzadeh, M. A.; Ollevier, T. Can. J. Chem. 2016, 94, 95–104. doi:10.1139/cjc-2015-0300 |
| 3. | Peighambardoust, S. J.; Rowshanzamir, S.; Amjadi, M. Int. J. Hydrogen Energy 2010, 35, 9349–9384. doi:10.1016/j.ijhydene.2010.05.017 |
| 40. | Asensio, J. A.; Sánchez, E. M.; Gómez-Romero, P. Chem. Soc. Rev. 2010, 39, 3210–3239. doi:10.1039/b922650h |
| 4. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 5. | Suzuki, A. J. Organomet. Chem. 1999, 576, 147–168. doi:10.1016/S0022-328X(98)01055-9 |
| 36. | Petrova, E.; Rasina, D.; Jirgensons, A. Eur. J. Org. Chem. 2017, 1773–1779. doi:10.1002/ejoc.201601582 |
| 9. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6 |
| 3. | Peighambardoust, S. J.; Rowshanzamir, S.; Amjadi, M. Int. J. Hydrogen Energy 2010, 35, 9349–9384. doi:10.1016/j.ijhydene.2010.05.017 |
| 9. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6 |
| 37. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Sokolenko, L. V.; Chernega, A. N.; Rusanov, E. B.; Tsymbal, I. F. J. Fluorine Chem. 2007, 128, 515–523. doi:10.1016/j.jfluchem.2007.01.002 |
| 38. | Yagupolskii, L. M.; Maletina, I. I.; Sokolenko, L. V.; Vlasenko, Y. G.; Buth, S. A. J. Fluorine Chem. 2008, 129, 486–492. doi:10.1016/j.jfluchem.2008.03.001 |
| 39. | Yagupolskii, L. M.; Maletina, I. I.; Sokolenko, L. V.; Vlasenko, Y. G.; Drozdova, M. V.; Polovinko, V. V. J. Fluorine Chem. 2010, 131, 238–247. doi:10.1016/j.jfluchem.2009.10.019 |
| 19. | Talanov, V. S.; Talanova, G. G.; Bartsch, R. A. Tetrahedron Lett. 2000, 41, 8221–8224. doi:10.1016/S0040-4039(00)01457-X |
| 20. | Talanov, V. S.; Talanova, G. G.; Gorbunova, M. G.; Bartsch, R. A. J. Chem. Soc., Perkin Trans. 2 2002, 209–215. doi:10.1039/b109638a |
| 21. | Talanov, V. S.; Talanova, G. G.; Gorbunova, M. G.; Bartsch, R. A. Tetrahedron Lett. 2002, 43, 1629–1631. doi:10.1016/S0040-4039(02)00096-5 |
| 22. | Surowiec, M.; Custelcean, R.; Surowiec, K.; Bartsch, R. A. Tetrahedron 2009, 65, 7777–7783. doi:10.1016/j.tet.2009.07.006 |
| 23. | Yang, Y.; Arora, G.; Fernandez, F. A.; Crawford, J. D.; Surowiec, K.; Lee, E. K.; Bartsch, R. A. Tetrahedron 2011, 67, 1389–1397. doi:10.1016/j.tet.2010.12.006 |
| 24. | Boston, A. L.; Lee, E. K.; Surowiec, K.; Gega, J.; Bartsch, R. A. Tetrahedron 2012, 68, 8789–8794. doi:10.1016/j.tet.2012.07.103 |
| 25. | Zhou, H.; Connery, K. E.; Bartsch, R. A.; Moyer, B. A.; Haverlock, T. J.; Delmau, L. H. Solvent Extr. Ion Exch. 2013, 31, 683–696. doi:10.1080/07366299.2013.806754 |
| 26. | Hendrickson, J. B.; Bergeron, R.; Giga, A.; Sternbach, D. J. Am. Chem. Soc. 1973, 95, 3412–3413. doi:10.1021/ja00791a072 |
| 27. | Hendrickson, J. B.; Bergeron, R. Tetrahedron Lett. 1973, 14, 4607–4610. doi:10.1016/S0040-4039(01)87289-0 |
| 28. | Benati, L.; Nanni, D.; Spagnolo, P. J. Org. Chem. 1999, 64, 5132–5138. doi:10.1021/jo9901541 |
| 29. | Pampín, M. C.; Estévez, J. C.; Estévez, R. J.; Maestro, M.; Castedo, L. Tetrahedron 2003, 59, 7231–7243. doi:10.1016/S0040-4020(03)01073-1 |
| 30. | Shainyan, B. A.; Tolstikova, L. L.; Bel'skikh, A. V. Russ. J. Org. Chem. 2008, 44, 1121–1125. doi:10.1134/S1070428008080022 |
| 31. | Moskalik, M. Y.; Meshcheryakov, V. I.; Shainyan, B. A. Russ. J. Org. Chem. 2009, 45, 1644–1650. doi:10.1134/S1070428009110116 |
| 32. | Cody, J. A.; Ahmed, I.; Tusch, D. J. Tetrahedron Lett. 2010, 51, 5585–5587. doi:10.1016/j.tetlet.2010.08.058 |
| 33. | Wu, C.; Li, P.; Shi, X.; Pan, X.; Wu, J. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o382. doi:10.1107/S1600536811001085 |
| 42. | Diness, F.; Begtrup, M. Org. Lett. 2014, 16, 3130–3133. doi:10.1021/ol5012554 |
| 15. | Lowe, J. A., III; Drozda, S. E.; McLean, S.; Bryce, D. K.; Crawford, R. T.; Zorn, S.; Morrone, J.; Appleton, T. A.; Lombardo, F. Bioorg. Med. Chem. Lett. 1995, 5, 1933–1936. doi:10.1016/0960-894X(95)00327-P |
| 16. | Hamada, Y.; Abdel-Rahman, H.; Yamani, A.; Nguyen, J.-T.; Stochaj, M.; Hidaka, K.; Kimura, T.; Hayashi, Y.; Saito, K.; Ishiurac, S.; Kiso, Y. Bioorg. Med. Chem. Lett. 2008, 18, 1649–1653. doi:10.1016/j.bmcl.2008.01.058 |
| 17. | Cervi, G.; Magnaghi, P.; Asa, D.; Avanzi, N.; Badari, A.; Borghi, D.; Caruso, M.; Cirla, A.; Cozzi, L.; Felder, E.; Galvani, A.; Gasparri, F.; Lomolino, A.; Magnuson, S.; Malgesini, B.; Motto, I.; Pasi, M.; Rizzi, S.; Salom, B.; Sorrentino, G.; Troiani, S.; Valsasina, B.; O’Brien, T.; Isacchi, A.; Donati, D.; D’Alessio, R. J. Med. Chem. 2014, 57, 10443–10454. doi:10.1021/jm501313x |
| 18. | Althaus, J.; Hake, T.; Hanekamp, W.; Lehr, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 131–140. doi:10.1080/14756366.2016.1178246 |
| 34. | Wu, X.; Rönn, R.; Gossas, T.; Larhed, M. J. Org. Chem. 2005, 70, 3094–3098. doi:10.1021/jo050080v |
| 35. | Urriolabeitia, E. P.; Laga, E.; Cativiela, C. Beilstein J. Org. Chem. 2012, 8, 1569–1575. doi:10.3762/bjoc.8.179 |
| 44. | Wang, J.-T.; Wainright, J. S.; Savinell, R. F.; Litt, M. J. Appl. Electrochem. 1996, 26, 751–756. doi:10.1007/bf00241516 |
| 12. | Leito, I.; Kaljurand, I.; Koppel, I. A.; Yagupolskii, L. M.; Vlasov, V. M. J. Org. Chem. 1998, 63, 7868–7874. doi:10.1021/jo981124l |
| 13. | Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; Pihl, V.; Koppel, I. A.; Leito, I. J. Phys. Org. Chem. 2013, 26, 162–170. doi:10.1002/poc.2946 |
| 14. | Trummal, A.; Lipping, L.; Kaljurand, I.; Koppel, I. A.; Leito, I. J. Phys. Chem. A 2016, 120, 3663–3669. doi:10.1021/acs.jpca.6b02253 |
| 41. | Diness, F.; Fairlie, D. P. Angew. Chem., Int. Ed. 2012, 51, 8012–8016. doi:10.1002/anie.201202149 |
| 42. | Diness, F.; Begtrup, M. Org. Lett. 2014, 16, 3130–3133. doi:10.1021/ol5012554 |
| 9. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6 |
| 10. | Yagupolskii, L. M.; Petrik, V. N.; Kondratenko, N. V.; Sooväli, L.; Kaljurand, I.; Leito, I.; Koppel, I. A. J. Chem. Soc., Perkin Trans. 2 2002, 1950–1955. doi:10.1039/B204172C |
| 11. | Koppel, I. A.; Burk, P.; Koppel, I.; Leito, I. J. Am. Chem. Soc. 2002, 124, 5594–5600. doi:10.1021/ja0255958 |
| 9. | Yagupolskii, L. M.; Shelyazhenko, S. V.; Maletina, I. I.; Petrik, V. N.; Rusanov, E. B.; Chernega, A. N. Eur. J. Org. Chem. 2001, 1225–1233. doi:10.1002/1099-0690(200104)2001:7<1225::AID-EJOC1225>3.0.CO;2-6 |
| 15. | Lowe, J. A., III; Drozda, S. E.; McLean, S.; Bryce, D. K.; Crawford, R. T.; Zorn, S.; Morrone, J.; Appleton, T. A.; Lombardo, F. Bioorg. Med. Chem. Lett. 1995, 5, 1933–1936. doi:10.1016/0960-894X(95)00327-P |
| 40. | Asensio, J. A.; Sánchez, E. M.; Gómez-Romero, P. Chem. Soc. Rev. 2010, 39, 3210–3239. doi:10.1039/b922650h |
© 2018 Diness et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)