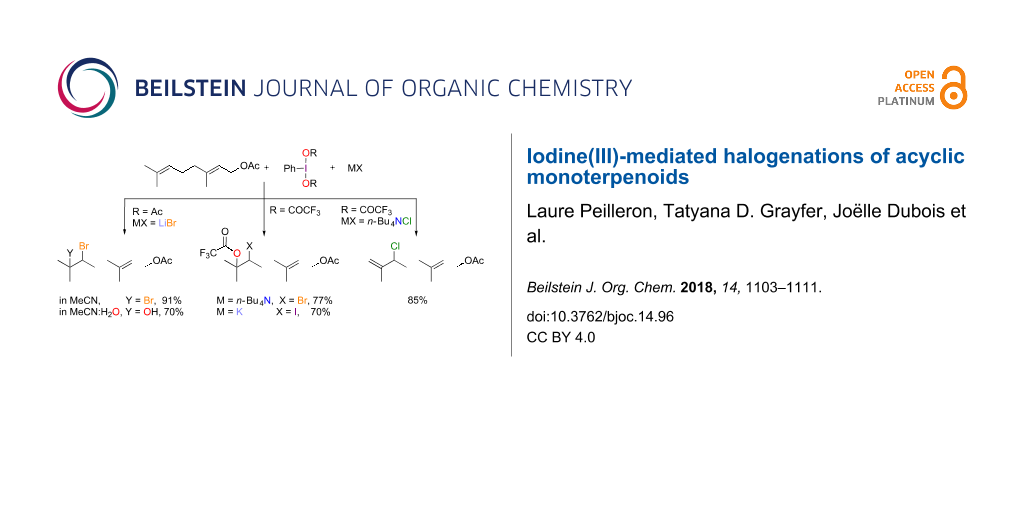
Abstract
Five different halofunctionalizations of acyclic monoterpenoids were performed using a combination of a hypervalent iodine(III) reagent and a halide salt. In this manner, the dibromination, the bromo(trifluoro)acetoxylation, the bromohydroxylation, the iodo(trifluoro)acetoxylation or the ene-type chlorination of the distal trisubstituted double bond occurred with excellent selectivity and moderate to good yields.

Graphical Abstract
Introduction
In nature, mostly in marine environments, halogenated compounds are produced by means of various enzymes that rely on widely available halides as feedstock [1]. These halogenases can perform an extremely wide array of electrophilic halogenations on a myriad of substrates with exquisite chemo-, stereo- and enantioselectivities that remain extremely challenging to rival for the chemist. When applied to terpenic feedstock, they give birth to a variety of molecular scaffolds, from the mono-chlorinated linear chain of fallachromenoic acid [2], to the pentahalogenated halomon skeleton [3] and encompassing many intricate polycyclic and macrocyclic structures with complex vicinal oxygen/halogen patterns, such as bromophycolide B [4] and dichotellide B, which contains both iodine and chlorine atoms (Figure 1) [5].
![[1860-5397-14-96-1]](/bjoc/content/figures/1860-5397-14-96-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Halogenated terpenoids from natural sources.
Figure 1: Halogenated terpenoids from natural sources.
This structural richness has fuelled the development of many synthetic strategies that take inspiration from these enzymatic machineries. A key aspect is to be able to mildly oxidize the halides into halenium equivalents in order to promote halogenations with increased selectivity. In this regard, hypervalent iodine reagents [6] have emerged as particularly versatile mediators [7-10]. We have shown that electrophilic halogenations [11-13], or pseudohalogenations [14] can be triggered by combining an iodine(III) derivative with a suitable halide salt. In particular, the chemoselectivity of the reaction can be finely tuned by adjusting several parameters, such as the nature of the halide as well as of the iodine(III) ligands and the halide counterion [15,16]. In the case of polyprenoids, we mostly devoted our efforts to achieve the bromocarbocyclization of aryl-geranyl derivatives using a combination of iodine(III) oxidant and a bromide source. In this fashion, the reaction of homogeranylbenzene with bis(tert-butylcarbonyloxy)iodobenzene and triethylsilyl bromide, followed by acidic treatment led to a tricyclic brominated adduct (Scheme 1, reaction 1). Yet in the course of our study we also showed that the reactivity of the key bridged bromonium intermediate could also be steered towards non-cyclizing vicinal difunctionalizations using slightly different combinations of a (diacyloxyiodo)arene and a bromide salt. Indeed, (diacetoxy)iodobenzene (DIB) and lithium bromide yield a dibromo adduct (Scheme 1, reaction 2), whereas a combination of (bis(trifluoroacetoxy)iodo)benzene (PIFA) and tetra-n-butylammonium bromide (TBAB) gives bromo(trifluoro)acetoxylated 3a (Scheme 1, reaction 3) [16].
![[1860-5397-14-96-i1]](/bjoc/content/inline/1860-5397-14-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previously developed bromo-functionalizations of polyprenoids using iodine(III) reagents.
Scheme 1: Previously developed bromo-functionalizations of polyprenoids using iodine(III) reagents.
We then decided to further explore the synthetic potential of the latter two vicinal difunctionalizations of terpenoids by not only expanding their substrate scope but also by trying to achieve other bromo-oxylations as well as analogous chloro- and/or iodofunctionalizations of linear terpenoids. For this purpose, five monoterpene derivatives – bearing various functional groups (protected and free alcohols, amine, E and Z olefins, diene moiety) aimed at probing potential chemo-, regio- and stereoselectivity issues – were selected: geranyl acetate (1a), neryl acetate (1b), geraniol (1c), N-tosylgeranylamine (1d) and myrcene (1e, Figure 2).
![[1860-5397-14-96-2]](/bjoc/content/figures/1860-5397-14-96-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected monoterpenoids used in this study.
Figure 2: Selected monoterpenoids used in this study.
Results and Discussion
Optimizations
In order to carry out the exploration of the various halogenations that could be performed, geranyl acetate (1a) was chosen as the model substrate. In our study on terpenoids [16], the reaction conditions for the dibromination of the distal double bond were easily established from our previous study on the bromination of enamides [11]. Thus, using a slight excess of DIB along with a two-fold amount of lithium bromide at 0 °C in dry acetonitrile rapidly yielded dibromo adduct 2a in 91% yield (Table 1, entry 1). Switching the reaction conditions to bromo(trifluoro)acetoxylation requires the use of a PIFA/TBAB combination in a 1:1 ratio with slow addition of the latter to the reaction mixture thereby preventing the formation of 2a. In this fashion, bromo(trifluoro)acetoxy adduct 3a was obtained in 77% yield (Table 1, entry 2). The reaction course can also be modified by changing the solvent. For instance, the complementary hydroxybromination reaction, giving 4a in 59% yield, was achievable if a mixture of acetonitrile and water was used as the solvent (Table 1, entry 3). In order to suppress the observed formation of minor amounts of 2a, the reaction temperature was lowered to −10 °C and the amount of lithium bromide, which was added dropwise as an aqueous solution, was diminished to 1.3 equivalents. By doing so, both the selectivity and the yield of 4a were improved though full conversion was not attained (Table 1, entry 4). Keeping the same procedure, complete reaction was nevertheless achieved by slightly increasing the amount of each reagent thus efficiently giving bromohydroxylated adduct 4a in 70% yield (Table 1, entry 5). Since the use of water as the sole solvent was not possible because of solubility issues, the reaction was attempted in ethanol. In this case the analogous bromoethoxylated adduct 4a’ could be isolated in 68% yield, albeit along with 25% of 2a (Table 1, entry 6). Turning our attention to iodination, we first used the combination of PIFA and KI that had given the best results with enamides [13]. Thus, iodo(trifluoro)acetoxylated adduct 5a was obtained in 70% yield (Table 1, entry 7). Interestingly, no traces of the diiodo compound were observed even if the hypervalent iodine(III) reagent was slowly added. Transposing the previously optimized bromo(trifluoro)acetoxylation conditions but using TBAI instead of KI did not improve the yield and reaction times and excess reagents were required to reach completion (Table 1, entry 8). Using directly 1.5 equivalents of both PIFA and TBAI did not suffice to improve the yield, whether in acetonitrile or in dichloromethane (Table 1, entries 9 and 10). When chlorination was attempted with a combination of DIB and iron(III) chloride in a 1.2:0.8 ratio (i.e., a 1:1 OAc/Cl ratio [12]) in acetonitrile, allylic chloride 6a [17] was obtained with a moderate 45% yield (Table 1, entry 11). Switching to a combination of PIFA and TBACl (Table 1, entry 12) did not change the course of the reaction towards chloro(trifluoro)acetoxylation and, while the yield was moderate in acetonitrile, it was greatly improved in dichloromethane [18], affording 6a in 85% yield (Table 1, entry 13).
Table 1: Optimization of the reactions conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-96-i8.svg?max-width=637&scale=1.0)
|
||||||
| entry | R (x equiv) | MX (y equiv) | solvent | temp. | time | N°, Y, X (yield %)a |
| 1 | Ac (1.2) | LiBr (2.4) | MeCN | 0 °C | 5 min | 2a, Br, Br (91) |
| 2 | C(O)CF3 (1.1) | n-Bu4NBrb (1.2) | MeCN | 0 °C | 15 minc | 3a, OCOCF3, Br (77) |
| 3 | Ac (1.2) | LiBr (2.4) | MeCN/H2O | rt | 5 min | 4a, OH, Br (59)d |
| 4 | Ac (1.2) | LiBre (1.3) | MeCN/H2O | −10 °C | 15 minc | 4a, OH, Br (65)f |
| 5 | Ac (1.4) | LiBre (1.6) | MeCN/H2O | −10 °C | 15 minc | 4a, OH, Br (70) |
| 6 | Ac (1.2) | LiBr (1.2) | EtOH | −10 °C | 105 min | 4a’, OEt, Br (68)g |
| 7 | C(O)CF3 (1.5)h | KI (2.4) | MeCN | 0 °C | 20 min | 5a, OCOCF3, I (70) |
| 8 | C(O)CF3 (1.3)i | n-Bu4NIb (1.5)i | MeCN | 0 °C | 100 min | 5a, OCOCF3, I (44) |
| 9 | C(O)CF3 (1.5) | n-Bu4NIb (1.5) | MeCN | 0 °C | 20 min | 5a, OCOCF3, I (60)j |
| 10 | C(O)CF3 (1.5) | n-Bu4NIb (1.5) | CH2Cl2 | 0 °C | 20 min | 5a, OCOCF3, I (63) |
| 11 | Ac (1.2) | FeCl3 (0.8) | MeCN | rt | 5 min | 6a (45) |
| 12 | C(O)CF3 (1.2) | n-Bu4NClb (1.5) | MeCN | 0 °C | 15 minc | 6a (36) |
| 13 | C(O)CF3 (1.2) | n-Bu4NClb (1.5) | CH2Cl2 | 0 °C | 15 minc | 6a (85) |
aIsolated yields; bslow addition of a 0.1 M solution of the TBA salt; c5 min of addition followed by 10 min of stirring; dalong with 6% of 2a; eslow addition of a 0.1 M aqueous solution of LiBr; ffull conversion was not reached; galong with 25% of 2a; hslow addition of a 0.1 M solution of PIFA; iinitially 1.1 equiv of PIFA and 1.2 equiv of TBAI, followed by 0.2 equiv of PIFA and 0.3 equiv of TBAI to reach completion; jalong with 20% of 1a.
At this stage, we chose to discard the bromoethoxylation reaction which led to mixtures of compounds, leaving five optimized transformations (dibromination, bromo(trifluoro)acetoxylation, bromohydroxylation, iodo(trifluoro)acetoxylation and ene-type chlorination) which were applied to substrates 1b–e in order to explore their scope.
Bromination
Dibromination
Application of the dibromination protocol to neryl acetate (1b) proceeded smoothly and the desired compound 2b was obtained in nearly quantitative yield without any detectable reaction on the Z double bond (Scheme 2). Despite the potential sensitivity of the alcohol and the amine functions, both geraniol (1c) and protected geranylamine 1d gave the dibromo adduct 2c [16] and 2d in good yields. Finally, the reaction was performed on myrcene (1e), providing 2e in 78% yield and leaving the diene moiety untouched.
![[1860-5397-14-96-i2]](/bjoc/content/inline/1860-5397-14-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Dibromination of acyclic monoterpenoids.
Scheme 2: Dibromination of acyclic monoterpenoids.
Bromo(trifluoro)acetoxylation
The success of the bromo(trifluoro)acetoxylation lies in the reverse addition protocol whereby the tetrabutylammonium bromide is slowly added to the suspension of the terpenoid and PIFA at 0 °C. In this manner, good to excellent yields (57–84%) were obtained for the formation of Z-derivative 3b, alcohol 3c [16], tosylamine 3d and diene 3e (Scheme 3). Once again, the selectivity is excellent as no reactions with the other double bonds or with the aryl ring [19] are observed and only one regioisomer is formed in all cases.
![[1860-5397-14-96-i3]](/bjoc/content/inline/1860-5397-14-96-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Bromo(trifluoro)acetoxylation of acyclic monoterpenoids.
Scheme 3: Bromo(trifluoro)acetoxylation of acyclic monoterpenoids.
Bromohydroxylation
Although the trifluoroacetoxy group can be readily cleaved [16], having a direct access to the corresponding bromohydrins would still be desirable especially since selective deprotection in the presence of another ester would presumably be difficult to achieve. By using water as the co-solvent, bromohydrins 4b–e were directly prepared from the corresponding starting material, albeit with moderate yields in the case of N-tosylgeranylamine (1d) and myrcene (1e, Scheme 4).
![[1860-5397-14-96-i4]](/bjoc/content/inline/1860-5397-14-96-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Bromohydroxylation of acyclic monoterpenoids.
Scheme 4: Bromohydroxylation of acyclic monoterpenoids.
Iodo(trifluoro)acetoxylation
In an analogous fashion to the above-described bromo(trifluoro)acetoxylation, the iodo(trifluoro)acetoxylation proceeded with both good yields (49–70%) and chemo-selectivities for geranyl and neryl acetates (1a,b), geranylamine 1d and myrcene (1e, Scheme 5). However, the adducts were quite sensitive and needed to be manipulated with care (low temperature storage) to avoid rapid decomposition. Nevertheless, this was particularly the case for alcohol 5c which could only be isolated with a 23% yield. This mild protocol offers a complementary alternative to the use of iodine monoacetate [20] or NIS [21] and does not require the use of a strong oxidant such as IBX [22]. Compared to the standard procedure for the preparation of acetoxyhypohalites that requires the use of expensive and potentially toxic silver salts [23], our method offers a more practical alternative. It also differs from the more user-friendly protocols that rely on Oxone® [20], DIB [24], or PIFA [25] since the iodide source is an iodide salt and not molecular iodine.
![[1860-5397-14-96-i5]](/bjoc/content/inline/1860-5397-14-96-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Iodo(trifluoro)acetoxylation of acyclic monoterpenoids.
Scheme 5: Iodo(trifluoro)acetoxylation of acyclic monoterpenoids.
Chlorination
In the case of chlorination, we have yet to observe adducts arising from the vicinal difunctionalization of the double bond and, in accordance with the literature [13], ene-type products 6 were obtained (Scheme 6). The reaction performed equally well with acetates (6a,b), alcohol (6c), amine (6d) and myrcene (6e). Once again, excellent chemo- and regioselectivities and good yields (51–85%) were witnessed.
![[1860-5397-14-96-i6]](/bjoc/content/inline/1860-5397-14-96-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Chlorination of acyclic monoterpenoids.
Scheme 6: Chlorination of acyclic monoterpenoids.
Mechanism proposal and control experiments
Considering the differences and similarities in the outcome of the various reaction conditions, a common mechanism with a central divergence point can be proposed. First, the diacetoxyiodobenzene reagent 7 would undergo ligand exchange with the halide to give mixed hypervalent iodine(III) 8 (Scheme 7a) [12]. We ruled out a possible direct reaction between the olefin and the hypervalent iodine(III) reagent as in the absence of any halide no reaction occurred within 1 h (Scheme 7b). A reductive elimination would then generate hypohalite R’OX which is presumably the active electrophilic species. If the reductive elimination only occurred after a second ligand exchange it would then give X2 which is generally a less efficient electrophilic species than the corresponding acetoxyhypohalite, especially trifluoroacetoxyhypohalites [26]. Indeed, when we reacted geranyl acetate 1a with bromine we did isolate dibromo adduct 2a but only with 47% yield and we also observed the formation of various byproducts such as ene-bromination adduct 6a’ (Scheme 7b) [27]. Moreover, for the iodination of enamides using the PIFA/KI combination we had already shown that the observed reactivity is more akin to the one induced by acetoxyhypoiodite [13]. Alternatively, the involvement of an ammonium-complexed halogen(I) species of type n-Bu4N[X(O2CCF3)2], as described by Kirschning [28] and later Muniz [29] cannot be ruled out although, to the best of our knowledge, it has yet to be characterized for X = Cl. Regardless of the actual halogenation species involved, regioselective halogenation of the terminal double bond of 1 would then give bridged halonium 9. From there three manifolds can be at play. Pathway a involves the addition of an oxygenated nucleophile. For X = Br, this is the case when the bromide is the default reagent because of its slow addition (giving 3) or because the nucleophile is the co-solvent (water giving 4 and to a lesser extent EtOH yielding 4’). While the formation of 4 is related to the well-established NBS-mediated formation of bromohydrins in aqueous media [30], the formation of 3 is not so straightforward. For instance, submitting 1a to NBS in the presence of trifluoroacetic acid led to a complex mixture of products, the major one being allylic bromide 6a’. For X = I, the steric bulk would preclude the formation of the vicinal diiodide and only pathway a would thus be operative, yielding 5. This is not the case for the dibromide 2, which is readily formed through pathway b when the reaction takes place in the presence of excess bromide. Finally, if X = Cl, rapid deprotonation by the acetate (or the chloride) alpha to the chloronium bridge (pathway c) explains the formation of allylic chloride 6.
![[1860-5397-14-96-i7]](/bjoc/content/inline/1860-5397-14-96-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: General mechanism proposal for the formation of 2–6 and control experiments.
Scheme 7: General mechanism proposal for the formation of 2–6 and control experiments.
Conclusion
Overall, we have further extended the scope of the iodine(III)-mediated oxidative halogenation of terpenoids which now includes dibromination, bromo(trifluoro)acetoxylation, bromohydroxylation, iodo(trifluoro)acetoxylation and allylic ene-chlorination. The conditions are mild and selective and the reactions proceed rapidly with generally good yields. Application of this tunable strategy towards the rapid constitution of chemical libraries of biologically active molecules is now actively pursued in our laboratory.
Experimental
General procedure A: dibromination
To a solution of the geranyl derivative (1.0 equiv) in acetonitrile (0.05 M) at 0 °C, lithium bromide (2.0–2.4 equiv), 4 Å molecular sieves (1.0 mass equiv) and DIB (1.2–1.4 equiv) were added. After stirring for 5 min, the reaction mixture was diluted with EtOAc, filtered over alumina (EtOAc) and concentrated under reduced pressure before purification by flash chromatography.
General procedure B: bromo(trifluoro)acetoxylation
To a solution of the geranyl derivative (1.0 equiv) in acetonitrile (0.07 M) cooled to 0 °C, PIFA (1.1 equiv) was added. A solution of tetra-n-butylammonium bromide (1.1 equiv) in acetonitrile (0.07 M) was then added dropwise over 5 min. After stirring for 10 min, the reaction mixture was diluted with EtOAc, saturated aqueous Na2S2O3 solution was added and the layers were separated. The aqueous layer was extracted twice with EtOAc. The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure before purification by flash chromatography.
General procedure C: bromohydroxylation
To a solution of the geranyl derivative (1.0 equiv) in acetonitrile (0.1 M) cooled to −10 °C, DIB (1.4 equiv) was added. A solution of lithium bromide (1.6 equiv) in H2O (0.1 M) was then added dropwise over 5 min. After stirring at −10 °C for 10 min, the reaction mixture was diluted with EtOAc, filtered over alumina (EtOAc) and concentrated under reduced pressure before purification by flash chromatography.
General procedure D: iodo(trifluoro)acetoxylation
To a solution of the geranyl derivative (1.0 equiv) in acetonitrile (0.1 M) at room temperature, 3 Å molecular sieves (1.0 mass equiv) and potassium iodide (2.4 equiv) were added. A solution of PIFA (1.5 equiv) in acetonitrile (0.1 M) was then added dropwise over 10 min and the reaction mixture was stirred for 15 min. The reaction mixture was diluted with EtOAc, Na2S2O3 solution (10%) was added and the layers were separated. The aqueous layer was extracted three times with EtOAc. The combined organic extracts were washed with water and brine, dried over MgSO4, filtered and concentrated under reduced pressure before purification by flash chromatography.
General procedure E: allylic chlorination
To a solution of the geranyl derivative (1.0 equiv) in dichloromethane (stabilized with amylene, 0.1 M) cooled to 0 °C, PIFA (1.2 equiv) was added. A solution of tetra-n-butylammonium chloride (1.5 equiv) in DCM (0.1 M) was then added dropwise over 10 min and the reaction mixture was stirred for 10 min at 0 °C. The reaction mixture was diluted with EtOAc, Na2S2O3 solution (10%) was added and the layers were separated. The aqueous layer was extracted three times with EtOAc. The combined organic extracts were washed with water and brine, dried over MgSO4, filtered and concentrated under reduced pressure before purification by flash chromatography.
Supporting Information
| Supporting Information File 1: Full characterization data of all new compounds and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Agarwal, V.; Miles, Z. D.; Winter, J. M.; Eustáquio, A. S.; El Gamal, A. A.; Moore, B. S. Chem. Rev. 2017, 117, 5619–5674. doi:10.1021/acs.chemrev.6b00571
Return to citation in text: [1] -
Reddy, P.; Urban, S. Phytochemistry 2009, 70, 250–255. doi:10.1016/j.phytochem.2008.12.007
Return to citation in text: [1] -
Sun, J.-F.; Huang, H.; Chai, X.-Y.; Yang, X.-W.; Meng, L.; Huang, C.-G.; Zhou, X.-F.; Yang, B.; Hu, J.; Chen, X.-Q.; Lei, H.; Wang, L.; Liu, Y. Tetrahedron 2011, 67, 1245–1250. doi:10.1016/j.tet.2010.11.087
Return to citation in text: [1] -
Kubanek, J.; Prusak, A. C.; Snell, T. W.; Giese, R. A.; Hardcastle, K. I.; Fairchild, C. R.; Aalbersberg, W.; Raventos-Suarez, C.; Hay, M. E. Org. Lett. 2005, 7, 5261–5264. doi:10.1021/ol052121f
Return to citation in text: [1] -
Burreson, J. B.; Woolard, F. X.; Moore, R. E. Chem. Lett. 1975, 4, 1111–1114. doi:10.1246/cl.1975.1111
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 424–431. doi:10.1055/s-0032-1318103
Return to citation in text: [1] -
Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582
Return to citation in text: [1] -
Martín Romero, R.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972–983. doi:10.1002/asia.201301637
Return to citation in text: [1] -
Arnold, A. M.; Ulmer, A.; Gulder, T. Chem. – Eur. J. 2016, 22, 8728–8739. doi:10.1002/chem.201600449
Return to citation in text: [1] -
Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R. H. Org. Lett. 2013, 15, 1842–1845. doi:10.1021/ol400453b
Return to citation in text: [1] [2] -
Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R. H. Tetrahedron 2014, 70, 6769–6775. doi:10.1016/j.tet.2014.07.066
Return to citation in text: [1] [2] [3] -
Beltran, R.; Nocquet-Thibault, S.; Blanchard, F.; Dodd, R. H.; Cariou, K. Org. Biomol. Chem. 2016, 14, 8448–8451. doi:10.1039/C6OB01673A
Return to citation in text: [1] [2] [3] [4] -
Nocquet-Thibault, S.; Rayar, A.; Retailleau, P.; Cariou, K.; Dodd, R. H. Chem. – Eur. J. 2015, 21, 14205–14210. doi:10.1002/chem.201501782
Return to citation in text: [1] -
Daniel, M.; Blanchard, F.; Nocquet-Thibault, S.; Cariou, K.; Dodd, R. H. J. Org. Chem. 2015, 80, 10624–10633. doi:10.1021/acs.joc.5b01750
Return to citation in text: [1] -
Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Demertzidou, V. P.; Pappa, S.; Sarli, V.; Zografos, A. L. J. Org. Chem. 2017, 82, 8710–8715. doi:10.1021/acs.joc.7b01103
Return to citation in text: [1] -
Hegde, S. G.; Vogel, M. K.; Saddler, J.; Hrinyo, T.; Rockwell, N.; Haynes, R.; Oliver, M.; Wolinsky, J. Tetrahedron Lett. 1980, 21, 441–444. doi:10.1016/S0040-4039(00)71427-4
Return to citation in text: [1] -
Zhou, Z.; He, X. Synthesis 2011, 207–209. doi:10.1055/s-0030-1258350
Return to citation in text: [1] -
Hokamp, T.; Storm, A. T.; Yusubov, M.; Wirth, T. Synlett 2018, 29, 415–418. doi:10.1055/s-0036-1589119
Return to citation in text: [1] [2] -
Smietana, M.; Gouverneur, V.; Mioskowski, C. Tetrahedron Lett. 2000, 41, 193–195. doi:10.1016/S0040-4039(99)02022-5
Return to citation in text: [1] -
Pan, Z.; Liu, X.; Liu, W.; Liang, Y. Synthesis 2005, 437–441. doi:10.1055/s-2004-834948
Return to citation in text: [1] -
Wilson, C. V. Org. React. 1957, 9, 332–362.
Return to citation in text: [1] -
Courtneidge, J. L.; Lusztyk, J.; Pagé, D. Tetrahedron Lett. 1994, 35, 1003–1006. doi:10.1016/S0040-4039(00)79950-3
Return to citation in text: [1] -
Merkushev, E. B.; Simakhina, N. D.; Koveshnikova, G. M. Synthesis 1980, 486–487. doi:10.1055/s-1980-29066
Return to citation in text: [1] -
Barnett, J. R.; Andrews, L. J.; Keefer, R. M. J. Am. Chem. Soc. 1972, 94, 6129–6134. doi:10.1021/ja00772a034
Return to citation in text: [1] -
Kato, T.; Ichinose, I. J. Chem. Soc., Perkin Trans. 1 1980, 1051–1056. doi:10.1039/P19800001051
Return to citation in text: [1] -
Monenschein, H.; Sourkouni-Argirusi, G.; Schubothe, K. M.; O’Hare, T.; Kirschning, A. Org. Lett. 1999, 1, 2101–2104. doi:10.1021/ol991149m
Return to citation in text: [1] -
Muñiz, K.; García, B.; Martínez, C.; Piccinelli, A. Chem. – Eur. J. 2017, 23, 1539–1545. doi:10.1002/chem.201603801
Return to citation in text: [1] -
Van Tamelen, E. E. Acc. Chem. Res. 1968, 1, 111–120. doi:10.1021/ar50004a003
Return to citation in text: [1]
| 25. | Merkushev, E. B.; Simakhina, N. D.; Koveshnikova, G. M. Synthesis 1980, 486–487. doi:10.1055/s-1980-29066 |
| 13. | Beltran, R.; Nocquet-Thibault, S.; Blanchard, F.; Dodd, R. H.; Cariou, K. Org. Biomol. Chem. 2016, 14, 8448–8451. doi:10.1039/C6OB01673A |
| 12. | Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R. H. Tetrahedron 2014, 70, 6769–6775. doi:10.1016/j.tet.2014.07.066 |
| 1. | Agarwal, V.; Miles, Z. D.; Winter, J. M.; Eustáquio, A. S.; El Gamal, A. A.; Moore, B. S. Chem. Rev. 2017, 117, 5619–5674. doi:10.1021/acs.chemrev.6b00571 |
| 5. | Burreson, J. B.; Woolard, F. X.; Moore, R. E. Chem. Lett. 1975, 4, 1111–1114. doi:10.1246/cl.1975.1111 |
| 12. | Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R. H. Tetrahedron 2014, 70, 6769–6775. doi:10.1016/j.tet.2014.07.066 |
| 4. | Kubanek, J.; Prusak, A. C.; Snell, T. W.; Giese, R. A.; Hardcastle, K. I.; Fairchild, C. R.; Aalbersberg, W.; Raventos-Suarez, C.; Hay, M. E. Org. Lett. 2005, 7, 5261–5264. doi:10.1021/ol052121f |
| 17. | Demertzidou, V. P.; Pappa, S.; Sarli, V.; Zografos, A. L. J. Org. Chem. 2017, 82, 8710–8715. doi:10.1021/acs.joc.7b01103 |
| 3. | Sun, J.-F.; Huang, H.; Chai, X.-Y.; Yang, X.-W.; Meng, L.; Huang, C.-G.; Zhou, X.-F.; Yang, B.; Hu, J.; Chen, X.-Q.; Lei, H.; Wang, L.; Liu, Y. Tetrahedron 2011, 67, 1245–1250. doi:10.1016/j.tet.2010.11.087 |
| 11. | Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R. H. Org. Lett. 2013, 15, 1842–1845. doi:10.1021/ol400453b |
| 29. | Muñiz, K.; García, B.; Martínez, C.; Piccinelli, A. Chem. – Eur. J. 2017, 23, 1539–1545. doi:10.1002/chem.201603801 |
| 2. | Reddy, P.; Urban, S. Phytochemistry 2009, 70, 250–255. doi:10.1016/j.phytochem.2008.12.007 |
| 13. | Beltran, R.; Nocquet-Thibault, S.; Blanchard, F.; Dodd, R. H.; Cariou, K. Org. Biomol. Chem. 2016, 14, 8448–8451. doi:10.1039/C6OB01673A |
| 14. | Nocquet-Thibault, S.; Rayar, A.; Retailleau, P.; Cariou, K.; Dodd, R. H. Chem. – Eur. J. 2015, 21, 14205–14210. doi:10.1002/chem.201501782 |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
| 13. | Beltran, R.; Nocquet-Thibault, S.; Blanchard, F.; Dodd, R. H.; Cariou, K. Org. Biomol. Chem. 2016, 14, 8448–8451. doi:10.1039/C6OB01673A |
| 11. | Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R. H. Org. Lett. 2013, 15, 1842–1845. doi:10.1021/ol400453b |
| 12. | Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R. H. Tetrahedron 2014, 70, 6769–6775. doi:10.1016/j.tet.2014.07.066 |
| 13. | Beltran, R.; Nocquet-Thibault, S.; Blanchard, F.; Dodd, R. H.; Cariou, K. Org. Biomol. Chem. 2016, 14, 8448–8451. doi:10.1039/C6OB01673A |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
| 28. | Monenschein, H.; Sourkouni-Argirusi, G.; Schubothe, K. M.; O’Hare, T.; Kirschning, A. Org. Lett. 1999, 1, 2101–2104. doi:10.1021/ol991149m |
| 7. | Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 424–431. doi:10.1055/s-0032-1318103 |
| 8. | Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582 |
| 9. | Martín Romero, R.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972–983. doi:10.1002/asia.201301637 |
| 10. | Arnold, A. M.; Ulmer, A.; Gulder, T. Chem. – Eur. J. 2016, 22, 8728–8739. doi:10.1002/chem.201600449 |
| 26. | Barnett, J. R.; Andrews, L. J.; Keefer, R. M. J. Am. Chem. Soc. 1972, 94, 6129–6134. doi:10.1021/ja00772a034 |
| 6. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 15. | Daniel, M.; Blanchard, F.; Nocquet-Thibault, S.; Cariou, K.; Dodd, R. H. J. Org. Chem. 2015, 80, 10624–10633. doi:10.1021/acs.joc.5b01750 |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
| 27. | Kato, T.; Ichinose, I. J. Chem. Soc., Perkin Trans. 1 1980, 1051–1056. doi:10.1039/P19800001051 |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
| 18. | Hegde, S. G.; Vogel, M. K.; Saddler, J.; Hrinyo, T.; Rockwell, N.; Haynes, R.; Oliver, M.; Wolinsky, J. Tetrahedron Lett. 1980, 21, 441–444. doi:10.1016/S0040-4039(00)71427-4 |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
| 20. | Hokamp, T.; Storm, A. T.; Yusubov, M.; Wirth, T. Synlett 2018, 29, 415–418. doi:10.1055/s-0036-1589119 |
| 24. | Courtneidge, J. L.; Lusztyk, J.; Pagé, D. Tetrahedron Lett. 1994, 35, 1003–1006. doi:10.1016/S0040-4039(00)79950-3 |
| 22. | Pan, Z.; Liu, X.; Liu, W.; Liang, Y. Synthesis 2005, 437–441. doi:10.1055/s-2004-834948 |
| 20. | Hokamp, T.; Storm, A. T.; Yusubov, M.; Wirth, T. Synlett 2018, 29, 415–418. doi:10.1055/s-0036-1589119 |
| 21. | Smietana, M.; Gouverneur, V.; Mioskowski, C. Tetrahedron Lett. 2000, 41, 193–195. doi:10.1016/S0040-4039(99)02022-5 |
| 16. | Grayfer, T. D.; Retailleau, P.; Dodd, R. H.; Dubois, J.; Cariou, K. Org. Lett. 2017, 19, 4766–4769. doi:10.1021/acs.orglett.7b02125 |
© 2018 Peilleron et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)