Abstract
Background: The carboxylesterase Notum has been shown to act as a key negative regulator of the Wnt signalling pathway by mediating the depalmitoleoylation of Wnt proteins. LP-922056 (1) is an orally active inhibitor of Notum. We are investigating the role of Notum in modulating Wnt signalling in the central nervous system and wished to establish if 1 would serve as a peripherally restricted control. An accessible and improved synthetic route would allow 1 to become more readily available as a chemical tool to explore the fundamental biology of Notum and build target validation to underpin new drug discovery programs.
Results: An improved, scalable synthesis of 1 is reported. Key modifications include: (1) the introduction of the C7-cyclopropyl group was most effectively achieved with a Suzuki–Miyaura cross-coupling reaction with MIDA-boronate 11 (5 → 6), and (2) C6 chlorination was performed with 1-chloro-1,2-benziodoxol-3-one (12) (6 → 7) as a mild and selective electrophilic chlorination agent. This 7-step route from 16 has been reliably performed on large scale to produce multigram quantities of 1 in good efficiency and high purity. Pharmacokinetic studies in mouse showed CNS penetration of 1 is very low with a brain/plasma concentration ratio of just 0.01. A small library of amides 17 were prepared from acid 1 to explore if 1 could be modified to deliver a CNS penetrant tool by capping off the acid as an amide. Although significant Notum inhibition activity could be achieved, none of these amides demonstrated the required combination of metabolic stability along with cell permeability without evidence of P-gp mediated efflux.
Conclusion: Mouse pharmacokinetic studies demonstrate that 1 is unsuitable for use in models of disease where brain penetration is an essential requirement of the compound but would be an ideal peripherally restricted control. These data will contribute to the understanding of drug levels of 1 to overlay with appropriate in vivo efficacy endpoints, i.e., the PK-PD relationship. The identification of a suitable analogue of 1 (or 17) which combines Notum inhibition with CNS penetration would be a valuable chemical probe for investigating the role of Notum in disease models.

Graphical Abstract
Introduction
The Wnt signalling pathway has been shown to regulate crucial aspects of cell fate determination, organogenesis, cell migration and polarity [1]. Importantly, compromised Wnt signalling has been implicated in the perturbation of synaptic integrity and function in Alzheimer’s disease (AD) [2]. Palmitoleoylation of Wnt proteins is required for efficient binding to Frizzled receptors and the subsequent signal transduction. The carboxylesterase Notum has been shown to act as a key negative regulator of the Wnt signalling pathway by specifically mediating the depalmitoleoylation of Wnt proteins [3,4].
LP-922056 (1, Figure 1) is an orally active inhibitor of Notum recently reported by Lexicon Pharmaceuticals [5,6]. Their research with 1 has shown that Notum is a potential drug target for stimulating bone formation and treating osteoporosis [7]. However, although 1 demonstrates low plasma clearance, the structure contains an essential carboxylic acid and acids tend to have low passive brain penetration [8-12]. We are investigating the role of Notum in modulating Wnt signalling in the central nervous system (CNS) [13] and wished to establish if 1 would serve as a peripherally restricted control compound. Hence, we required a synthetic route to 1 that could be reliably and safely performed on large scale.
![[1860-5397-15-271-1]](/bjoc/content/figures/1860-5397-15-271-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of Notum inhibitor LP-922056 (1).
Figure 1: Chemical structure of Notum inhibitor LP-922056 (1).
A synthesis of 1 has been published in the patent literature [6], although many of the experimental procedures are described in terms of ‘general procedures’ which do not seem to work well when applied to 1 which contains functional groups sensitive to certain reagents employed (vide infra). An improved synthetic route should allow 1 to become more readily available as a chemical tool to explore the fundamental biology of Notum and build target validation to underpin new drug discovery programs for non-CNS disease.
Results and Discussion
Improved synthesis of 1; first generation
Our first complete synthesis of 1 is presented in Scheme 1 (see Supporting Information File 1 for experimental procedures and characterisation data).
![[1860-5397-15-271-i1]](/bjoc/content/inline/1860-5397-15-271-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
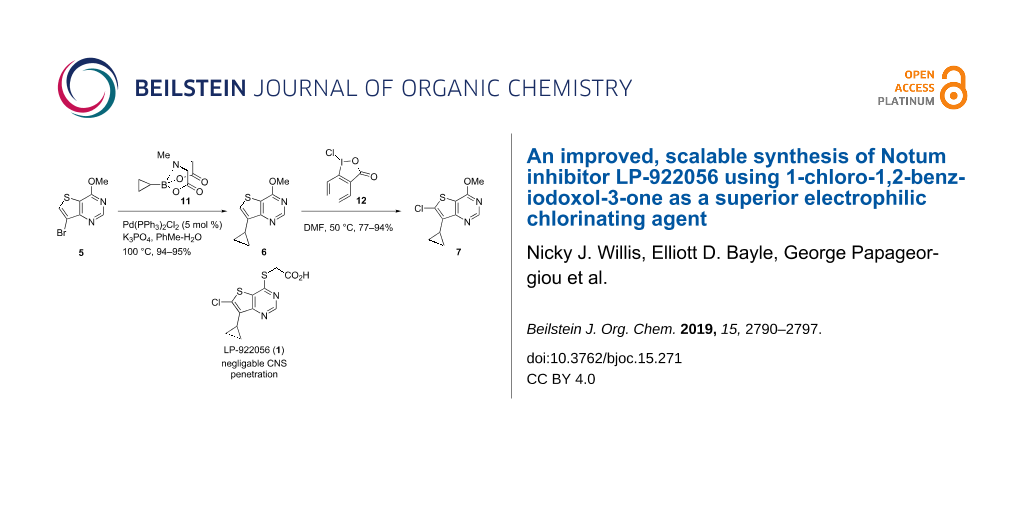
Scheme 1: Synthesis of LP-922056 (1). Reagents and conditionsa: (a) (COCl)2 (3.3 equiv), DMF, CH2Cl2, 55 °C , 16 h, 63–78%; (b) NaOMe (5 equiv), 1,4-dioxane, 0 °C then rt, 16 h, 92–93%; (c) NBS (1.1 equiv), AcOH/MeCN (1:100), 85 °C, 16 h, 41–48%; (d) 11 (1.5 equiv), Pd(PPh3)2Cl2 (5 mol %), K3PO4 (6 equiv), PhMe/H2O (3:1, 0.25 M), 100 °C, 16 h, 94–95%; (e) 1-chloro-1,2-benziodoxol-3-one (12, 1.5 equiv), DMF, 50 °C, 16 h, 77–94%; (f) HCl (12 M, 40 equiv), 70 °C, 16 h; (g) POCl3 (20 equiv), 90 °C, 16 h, 81% over 2 steps; (h) HSCH2CO2Me (13, 1.2 equiv), NEt3 (2.1 equiv), MeOH, 0 °C to rt, 16 h, 84%; (i) NaOH (1 M, 2 equiv), THF, 0 °C, 1 h, then HCl (1 M), 0 °C, 30 min, 98%. aThese reactions have been performed several times but have not been systematically optimised. Yields are the ranges obtained from repeated reactions. DMF, N,N-dimethylformamide; NBS, N-bromosuccinimide; THF, tetrahydrofuran.
Scheme 1: Synthesis of LP-922056 (1). Reagents and conditionsa: (a) (COCl)2 (3.3 equiv), DMF, CH2Cl2, 55 °C ,...
This short sequence starts with 4-chlorothieno[3,2-d]pyrimidine (3), which is readily available from commercial suppliers, and generally follows published procedures [5,6] but with key modifications to increase yields/selectivities and significantly improve ease of purification of key intermediates. Our modifications include: (1) the introduction of the C7-cyclopropyl group was most effectively achieved with a Suzuki–Miyaura cross-coupling reaction with MIDA-boronate 11 (5 → 6); and (2) C6 chlorination was performed with 1-chloro-1,2-benziodoxol-3-one (12) (6 → 7) as a mild selective electrophilic chlorination agent.
4-Chlorothieno[3,2-d]pyrimidine (3) was either purchased or prepared from thieno[3,2-d]pyrimidin-4(3H)-one (2) by C4 chlorination with oxalyl chloride/DMF following the method of Mitchell et al. [14]. Treatment of 3 with NaOMe displaced the C4–Cl to give 4 in a good yield as described by Atheral et al. [15]. Thieno[3,2-d]pyrimidine 4 is now suitably functionalised for the introduction of the C7-cyclopropyl group, the C6-chlorine atom and elaboration of the thioacetic acid moiety at C4.
Electrophilic bromination at C7 with N-bromosuccinimide gave 5 as the major regioisomer reproducibly on 100 mmol scale in modest yield (41–48%). This proved to be the least efficient step in our sequence and justified further optimisation (vide infra). Suzuki–Miyaura cross coupling of bromide 5 with cyclopropylboronic acid (2.5 equiv) produced 6 in good yield (62–89%) but the product required extensive chromatographic purification. We reasoned that switching from the boronic acid (c-PrB(OH)2) to the corresponding MIDA-boronate 11 would improve the quality of the reagent and slow release of the active boron species during the course of the reaction would allow us to reduce the number of molar equivalents required to improve conversion [16]. Palladium-mediated cross coupling of 5 with 11 (1.5 equiv) gave 6 in reproducibly high yield (ca. 95%) when performed on gram scale. However, when performed on larger scale (9.3 g), the reaction stalled after ca. 90% conversion and addition of extra catalyst Pd[(PPh3)2Cl2] and/or 11 failed to drive the reaction to completion. We found the most efficient way to complete the reaction conversion was to isolate the crude product (mostly 6) and subject this material to a repeat reaction; this procedure gave 6 in a good yield 95% and simplified purification.
With multigram quantities of 6 in hand, attention was turned to the C6 chlorination step. Unfortunately, despite this reaction being reported in the literature, there were no experimental details for this specific transformation as only a ‘general procedure’ was described [6]. Our attempts to use this procedure with N-chlorosuccinimide (NCS) as the chlorinating agent gave poor yields of the desired product 7 (ca. 15–32%) due to competing ring opening reactions of the 7-cyclopropyl group (Scheme 2). Clearly, a better procedure was required.
![[1860-5397-15-271-i2]](/bjoc/content/inline/1860-5397-15-271-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chlorination of 6 with N-chlorosuccinimide (NCS). Reagents and conditions: (a) NCS (1.2 equiv), AcOH, 55 °C, 7 h, 15–32%.
Scheme 2: Chlorination of 6 with N-chlorosuccinimide (NCS). Reagents and conditions: (a) NCS (1.2 equiv), AcO...
A large number of electrophilic chlorinating reagents for the direct chlorination of aromatic rings have been reported [17]. Recently, Xue et al. described the electrophilic chlorination of arenes and heterocycles by 1-chloro-1,2-benziodoxol-3-one (12) [18,19]. The hypervalent iodine(III) reagent 12 is reported to be a mild and effective reagent for the chlorination of nitrogen containing heterocycles which is easy to prepare and is air- and moisture-stable. The scope of published substrates includes chlorination of 7H-pyrrolo[2,3-d]pyrimidines and we wished to see if we could extend the scope to include sulphur containing heterocycles such as thieno[3,2-d]pyrimidines (e.g., 6). It was also important to explore if 12 would efficiently chlorinate 6 at the less activated C6 position in the presence of the C7 cyclopropyl group.
Treatment of 6 with 12 (1.5 equiv) in DMF at 50 °C for 16–24 h gave the desired chloro product 7 in 77–94% isolated yield. Analysis of the crude reaction mixture showed only trace amounts of cyclopropyl ring opened products such as 14/15 as detectible by LC–MS. Hence, 12 proved to be a far superior reagent, when compared to NCS, for the C6 chlorination of thieno[3,2-d]pyrimidine 6 (i.e., 6 → 7).
Completion of our synthesis of 1 followed established procedures although it proved expedient to carry material through several of these later steps without the need for extensive purification beyond a simple work-up procedure (7 → 8 → 9). Activation of C4 was accomplished by a two-step procedure of acid hydrolysis of the C4-OMe of 7 to give thieno[3,2-d]pyrimidin-4(3H)-one 8, followed by chlorination with POCl3 to give 9. Finally, nucleophilic displacement of the C4–Cl of 9 by methyl thioglycolate (13) gave ester 10 which was hydrolysed with NaOH to afford 1. This route has been reliably performed on large scale to produce multigram quantities of 1 in good efficiency (total yield over 8-steps from 3: 18–26%) and high purity (>99%).
Improved synthesis of 1; second generation
A shorter synthesis was then developed by accessing bromide 5 by an alternative route. The low-yielding C7 bromination of 4 with NBS to give 5 as described above (Scheme 1, step c) was avoided by starting with 7-bromo-4-chlorothieno[3,2-d]pyrimidine (16) which is readily available from commercial suppliers. Treatment of 16 with NaOMe displaced the C4–Cl to give 5 in good yield on 10 g scale (Scheme 3). Even though 16 is somewhat more expensive than 2 or 3 per unit cost (by ca. 5-fold), this updated route shortens the sequence to just 7 steps from 16 and improves the overall yield to 40–50 %.
![[1860-5397-15-271-i3]](/bjoc/content/inline/1860-5397-15-271-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Improved synthesis of 5. Reagents and conditions: (a) NaOMe (5 equiv), 1,4-dioxane, 0 °C then rt, 16 h, 84%.
Scheme 3: Improved synthesis of 5. Reagents and conditions: (a) NaOMe (5 equiv), 1,4-dioxane, 0 °C then rt, 1...
Mouse pharmacokinetics for 1
Assessment of 1 in mouse liver microsomes (MLM) showed excellent metabolic stability (Cli 1.0 μL/min/mg protein) which predicts for low clearance in vivo. Binding to mouse plasma proteins (mPPB) was very high with percent unbound drug (fu) of just 0.1%; this mPPB value can be used to calculate free drug concentrations from measured drug levels in plasma taken during in vivo experiments. The high mPPB is entirely consistent with the physicochemical properties of 1 as a lipophilic acid (mw 300; cLogP 3.1; cpKa 3.1).
Pharmacokinetic (PK) data for 1 was generated in vivo in mouse to evaluate brain penetration (Table 1; Figure 2) (see Supporting Information File 1, Tables S1–S3). The route of administration and dose were selected to most closely match relevant published mouse disease model studies [5,7]. Following single oral dose (p.o.) of 10 mg/kg, plasma exposure was high and plasma clearance was low relative to liver blood flow resulting in a plasma elimination half-life of 8.8 hours. The plasma parameters from these mouse PK experiments (Cmax and AUC) are consistent with published preclinical PK data [5].
Table 1: Mouse pharmacokinetic data for 1; oral (p.o.) dose at 10 mg/kg.a
| PK Parameter | Plasma | Brain |
| T1/2 | 8.8 h | 7.1 h |
| Tmax | 2.0 h | 2.0 h |
| Cmax | 35,400 ng/mL | 500 ng/g |
| AUC0→t | 303,000 h·ng/mL | 3,700 h·ng/g |
| AUC0→∞ | 354,000 h·ng/mL | 4,080 h·ng/g |
aMale fed CD1 mouse; suspension formulation in 0.1% Tween80 in water; n = 3 per time point; terminal blood and brain levels measured at seven time points: 0.17, 0.50, 1, 2, 5, 7.5 and 24 h.
![[1860-5397-15-271-2]](/bjoc/content/figures/1860-5397-15-271-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Concentrations of 1 in mouse following oral administration (p.o.) at 10 mg/kg.
Figure 2: Concentrations of 1 in mouse following oral administration (p.o.) at 10 mg/kg.
CNS penetration of 1 was very low with a brain/plasma concentration ratio of ≈0.01 at all time points measured and was also 0.01 based on AUC(0→∞). At this level of exposure, a significant proportion of the compound detected in brain samples is likely to have arisen from residual blood in the brain tissue.
Amide analogues of 1 to explore CNS penetration and future opportunities
With a peripherally restricted control in hand, we elected to explore if 1 could be modified to deliver a CNS penetrant tool by capping off the acid as an amide. A small library of amides 17 were prepared from acid 1 by activation with HBTU and then subsequent reaction with the amine 18 (Scheme 4). These amides 17 were designed to have molecular properties (mw, cLogP, tPSA, HBD, pKa) consistent with CNS drug-like space [20].
![[1860-5397-15-271-i4]](/bjoc/content/inline/1860-5397-15-271-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of amides 17. Representative reagents and conditionsa: (a) HBTU (1.1 equiv), iPr2NEt (2.5 equiv), DMF, rt, 15 min; then amine 18 (1.05 equiv). aThese reactions were performed once for each amide and have not been optimised. DMF, N,N-dimethylformamide; HBTU, O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate.
Scheme 4: Preparation of amides 17. Representative reagents and conditionsa: (a) HBTU (1.1 equiv), iPr2NEt (2...
Although significant Notum inhibition activity could be achieved (IC50 < 100 nM), none of these specific amides demonstrated the required combination of sufficient MLM stability along with cell permeability as measured by transit performance across a MDCK-MDR1 monolayer without evidence of P-gp mediated efflux. This collection of data for amides 17a–r is shared as ‘open data’ to assist others in evaluating these results with the objective of solving this challenge (see Supporting Information File 1, Table S4 and Supporting Information File 2). Our own efforts took us to an alternative chemotype [21].
Conclusion
An improved, scalable synthesis of Notum inhibitor 1 is reported. Key modifications include: (1) the introduction of the C7-cyclopropyl group was most effectively achieved with a Suzuki–Miyaura cross-coupling reaction with MIDA-boronate 11 (5 → 6); and (2) C6 chlorination was performed with 1-chloro-1,2-benziodoxol-3-one (12) (6 → 7) as a mild selective electrophilic chlorination agent. This 7-step route from 16 has been reliably performed on large scale to produce multigram quantities of 1 in good efficiency.
Pharmacokinetic studies in mouse showed CNS penetration of 1 is very low with brain/plasma concentration ratio of just 0.01 based on AUC(0→∞). Hence, 1 is unsuitable for use in models of disease where brain penetration is an essential requirement of the compound but would be an ideal peripherally restricted control. These data will contribute to the understanding of drug levels of 1 to overlay with appropriate in vivo efficacy endpoints, i.e., the PK-PD relationship. The full PK data set is presented and shared as ‘open data’. This complete data set (along with others) will also assist with the creation of improved predictive pharmacokinetic models.
A small library of amides 17 were prepared from acid 1 to explore if 1 could be modified to deliver a CNS penetrant tool by capping off the acid as an amide. Although significant Notum inhibition activity could be achieved, none of these amides demonstrated the required combination of metabolic stability along with cell permeability without evidence of P-gp mediated efflux. The identification of a suitable analogue of 1 (or 17) which combines Notum inhibition with CNS penetration would be a valuable chemical probe for investigating the role of Notum in disease models. This collection of data for amides 17a–r is shared as ‘open data’ to assist others in evaluating these results with the objective of solving this challenge.
Supporting Information
(1) experimental procedures and characterisation data for 3–10 and 1; (2) mouse PK data which includes: study design summary; plasma concentrations; brain concentrations; (3) Notum IC50 (nM), MLM Cli (μL/min/mg protein) and MDCK-MDR1 AB/BA Papp (× 10−6 cm/s) data for 17a–r.
To view the NMR spectra, use the file within the pdata folder of Supporting Information File 3.
| Supporting Information File 1: Experimental section, mouse pharmacokinetics and profiles of amides. | ||
| Format: PDF | Size: 905.8 KB | Download |
| Supporting Information File 2: Profiles of amides. | ||
| Format: CSV | Size: 1.3 KB | Download |
| Supporting Information File 3: Raw NMR data files for compound LP-922056. | ||
| Format: ZIP | Size: 2.4 MB | Download |
Acknowledgements
This work was supported by Alzheimer’s Research UK (ARUK) and The Francis Crick Institute. The ARUK UCL Drug Discovery Institute is core funded by Alzheimer’s Research UK (520909). The Francis Crick Institute receives its core funding from Cancer Research UK (FC001002), the UK Medical Research Council (FC001002), and the Wellcome Trust (FC001002).
We thank our colleagues Sarah Frew, Amy Monaghan, Fiona Jeganathan and Magda Bictash of the ARUK UCL DDI Screening and Pharmacology team for Notum inhibition data. We thank Abil Aliev and Kersti Karu at the UCL Department of Chemistry for spectroscopic and analytical services. Mouse PK studies were performed by Pharmidex (London, UK) and ADME studies reported in this work were performed by GVK Biosciences (Hyderabad, India) and Cyprotex (Macclesfield, UK).
References
-
Komiya, Y.; Habas, R. Organogenesis 2008, 4, 68–75. doi:10.4161/org.4.2.5851
Return to citation in text: [1] -
Palomer, E.; Buechler, J.; Salinas, P. C. Front. Cell. Neurosci. 2019, 13, No. 227. doi:10.3389/fncel.2019.00227
Return to citation in text: [1] -
Kakugawa, S.; Langton, P. F.; Zebisch, M.; Howell, S. A.; Chang, T.-H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A. P.; Jones, E. Y.; Vincent, J.-P. Nature 2015, 519, 187–192. doi:10.1038/nature14259
Return to citation in text: [1] -
Zhang, X.; Cheong, S.-M.; Amado, N. G.; Reis, A. H.; MacDonald, B. T.; Zebisch, M.; Jones, E. Y.; Abreu, J. G.; He, X. Dev. Cell 2015, 32, 719–730. doi:10.1016/j.devcel.2015.02.014
Return to citation in text: [1] -
Tarver, J. E., Jr.; Pabba, P. K.; Barbosa, J.; Han, Q.; Gardyan, M. W.; Brommage, R.; Thompson, A. Y.; Schmidt, J. M.; Wilson, A. G. E.; He, W.; Lombardo, V. K.; Carson, K. G. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. doi:10.1016/j.bmcl.2016.02.021
Return to citation in text: [1] [2] [3] [4] -
Barbosa, J.; Carson, K. G.; Gardyan, M. W.; He, W.; Lombardo, V.; Pabba, P.; Tarver, J., Jr. Inhibitors of Notum pectinacylesterase and methods of their use. U.S. Pat. Appl. US 2012/0065200 A1, March 15, 2012.
Return to citation in text: [1] [2] [3] [4] -
Brommage, R.; Liu, J.; Vogel, P.; Mseeh, F.; Thompson, A. Y.; Potter, D. G.; Shadoan, M. K.; Hansen, G. M.; Jeter-Jones, S.; Cui, J.; Bright, D.; Bardenhagen, J. P.; Doree, D. D.; Movérare-Skrtic, S.; Nilsson, K. H.; Henning, P.; Lerner, U. H.; Ohlsson, C.; Sands, A. T.; Tarver, J. E.; Powell, D. R.; Zambrowicz, B.; Liu, Q. Bone Res. 2019, 7, 2. doi:10.1038/s41413-018-0038-3
Return to citation in text: [1] [2] -
Manallack, D. T. Perspect. Med. Chem. 2007, 1, 25–38. doi:10.1177/1177391x0700100003
Return to citation in text: [1] -
Wager, T. T.; Chandrasekaran, R. Y.; Hou, X.; Troutman, M. D.; Verhoest, P. R.; Villalobos, A.; Will, Y. ACS Chem. Neurosci. 2010, 1, 420–434. doi:10.1021/cn100007x
Return to citation in text: [1] -
Manallack, D. T.; Prankerd, R. J.; Yuriev, E.; Oprea, T. I.; Chalmers, D. K. Chem. Soc. Rev. 2013, 42, 485–496. doi:10.1039/c2cs35348b
Return to citation in text: [1] -
Charifson, P. S.; Walters, W. P. J. Med. Chem. 2014, 57, 9701–9717. doi:10.1021/jm501000a
Return to citation in text: [1] -
Di, L.; Rong, H.; Feng, B. J. Med. Chem. 2013, 56, 2–12. doi:10.1021/jm301297f
Return to citation in text: [1] -
Atkinson, B. N.; Steadman, D.; Zhao, Y.; Sipthorp, J.; Vecchia, L.; Ruza, R. R.; Jeganathan, F.; Lines, G.; Frew, S.; Monaghan, A.; Kjær, S.; Bictash, M.; Jones, E. Y.; Fish, P. V. Med. Chem. Commun. 2019, 10, 1361–1369. doi:10.1039/c9md00096h
Return to citation in text: [1] -
Mitchell, I. S.; Spencer, K. L.; Stengel, P.; Han, Y.; Kallan, N. C.; Munson, M.; Vigers, G. P. A.; Blake, J.; Piscopio, A.; Josey, J.; Miller, S.; Xiao, D.; Xu, R.; Rao, C.; Wang, B.; Bernacki, A. L. Akt protein kinase inhibitors. WO Patent WO2005/051304, June 9, 2005.
Return to citation in text: [1] -
Atheral, J. F.; Hough, T. L.; Lindell, S. D.; O'Mahony, M. J.; Parsons, J. H.; Saville-Stones, E. A. Fungicides. U.S. Patent US6432964B1, Aug 13, 2002.
Return to citation in text: [1] -
Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204
Return to citation in text: [1] -
Rogers, D. A.; Bensalah, A. T.; Espinosa, A. T.; Hoerr, J. L.; Refai, F. H.; Pitzel, A. K.; Alvarado, J. J.; Lamar, A. A. Org. Lett. 2019, 21, 4229–4233. doi:10.1021/acs.orglett.9b01414
Return to citation in text: [1] -
Wang, M.; Zhang, Y.; Wang, T.; Wang, C.; Xue, D.; Xiao, J. Org. Lett. 2016, 18, 1976–1979. doi:10.1021/acs.orglett.6b00547
Return to citation in text: [1] -
Matoušek, V.; Pietrasiak, E.; Schwenk, R.; Togni, A. J. Org. Chem. 2013, 78, 6763–6768. doi:10.1021/jo400774u
Return to citation in text: [1] -
Rankovic, Z. J. Med. Chem. 2015, 58, 2584–2608. doi:10.1021/jm501535r
Return to citation in text: [1] -
Fish, P. V. Development of Potent, Selective, CNS Penetrant Small Molecule Inhibitors of Notum to Potentiate Wnt Signalling. Presented at EFMC: XXVth International Symposium on Medicinal Chemistry; 2–6th September 2018; Ljubljana, Slovenia.
Return to citation in text: [1]
| 21. | Fish, P. V. Development of Potent, Selective, CNS Penetrant Small Molecule Inhibitors of Notum to Potentiate Wnt Signalling. Presented at EFMC: XXVth International Symposium on Medicinal Chemistry; 2–6th September 2018; Ljubljana, Slovenia. |
| 5. | Tarver, J. E., Jr.; Pabba, P. K.; Barbosa, J.; Han, Q.; Gardyan, M. W.; Brommage, R.; Thompson, A. Y.; Schmidt, J. M.; Wilson, A. G. E.; He, W.; Lombardo, V. K.; Carson, K. G. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. doi:10.1016/j.bmcl.2016.02.021 |
| 7. | Brommage, R.; Liu, J.; Vogel, P.; Mseeh, F.; Thompson, A. Y.; Potter, D. G.; Shadoan, M. K.; Hansen, G. M.; Jeter-Jones, S.; Cui, J.; Bright, D.; Bardenhagen, J. P.; Doree, D. D.; Movérare-Skrtic, S.; Nilsson, K. H.; Henning, P.; Lerner, U. H.; Ohlsson, C.; Sands, A. T.; Tarver, J. E.; Powell, D. R.; Zambrowicz, B.; Liu, Q. Bone Res. 2019, 7, 2. doi:10.1038/s41413-018-0038-3 |
| 18. | Wang, M.; Zhang, Y.; Wang, T.; Wang, C.; Xue, D.; Xiao, J. Org. Lett. 2016, 18, 1976–1979. doi:10.1021/acs.orglett.6b00547 |
| 19. | Matoušek, V.; Pietrasiak, E.; Schwenk, R.; Togni, A. J. Org. Chem. 2013, 78, 6763–6768. doi:10.1021/jo400774u |
| 5. | Tarver, J. E., Jr.; Pabba, P. K.; Barbosa, J.; Han, Q.; Gardyan, M. W.; Brommage, R.; Thompson, A. Y.; Schmidt, J. M.; Wilson, A. G. E.; He, W.; Lombardo, V. K.; Carson, K. G. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. doi:10.1016/j.bmcl.2016.02.021 |
| 6. | Barbosa, J.; Carson, K. G.; Gardyan, M. W.; He, W.; Lombardo, V.; Pabba, P.; Tarver, J., Jr. Inhibitors of Notum pectinacylesterase and methods of their use. U.S. Pat. Appl. US 2012/0065200 A1, March 15, 2012. |
| 5. | Tarver, J. E., Jr.; Pabba, P. K.; Barbosa, J.; Han, Q.; Gardyan, M. W.; Brommage, R.; Thompson, A. Y.; Schmidt, J. M.; Wilson, A. G. E.; He, W.; Lombardo, V. K.; Carson, K. G. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. doi:10.1016/j.bmcl.2016.02.021 |
| 7. | Brommage, R.; Liu, J.; Vogel, P.; Mseeh, F.; Thompson, A. Y.; Potter, D. G.; Shadoan, M. K.; Hansen, G. M.; Jeter-Jones, S.; Cui, J.; Bright, D.; Bardenhagen, J. P.; Doree, D. D.; Movérare-Skrtic, S.; Nilsson, K. H.; Henning, P.; Lerner, U. H.; Ohlsson, C.; Sands, A. T.; Tarver, J. E.; Powell, D. R.; Zambrowicz, B.; Liu, Q. Bone Res. 2019, 7, 2. doi:10.1038/s41413-018-0038-3 |
| 3. | Kakugawa, S.; Langton, P. F.; Zebisch, M.; Howell, S. A.; Chang, T.-H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A. P.; Jones, E. Y.; Vincent, J.-P. Nature 2015, 519, 187–192. doi:10.1038/nature14259 |
| 4. | Zhang, X.; Cheong, S.-M.; Amado, N. G.; Reis, A. H.; MacDonald, B. T.; Zebisch, M.; Jones, E. Y.; Abreu, J. G.; He, X. Dev. Cell 2015, 32, 719–730. doi:10.1016/j.devcel.2015.02.014 |
| 6. | Barbosa, J.; Carson, K. G.; Gardyan, M. W.; He, W.; Lombardo, V.; Pabba, P.; Tarver, J., Jr. Inhibitors of Notum pectinacylesterase and methods of their use. U.S. Pat. Appl. US 2012/0065200 A1, March 15, 2012. |
| 2. | Palomer, E.; Buechler, J.; Salinas, P. C. Front. Cell. Neurosci. 2019, 13, No. 227. doi:10.3389/fncel.2019.00227 |
| 17. | Rogers, D. A.; Bensalah, A. T.; Espinosa, A. T.; Hoerr, J. L.; Refai, F. H.; Pitzel, A. K.; Alvarado, J. J.; Lamar, A. A. Org. Lett. 2019, 21, 4229–4233. doi:10.1021/acs.orglett.9b01414 |
| 5. | Tarver, J. E., Jr.; Pabba, P. K.; Barbosa, J.; Han, Q.; Gardyan, M. W.; Brommage, R.; Thompson, A. Y.; Schmidt, J. M.; Wilson, A. G. E.; He, W.; Lombardo, V. K.; Carson, K. G. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. doi:10.1016/j.bmcl.2016.02.021 |
| 6. | Barbosa, J.; Carson, K. G.; Gardyan, M. W.; He, W.; Lombardo, V.; Pabba, P.; Tarver, J., Jr. Inhibitors of Notum pectinacylesterase and methods of their use. U.S. Pat. Appl. US 2012/0065200 A1, March 15, 2012. |
| 15. | Atheral, J. F.; Hough, T. L.; Lindell, S. D.; O'Mahony, M. J.; Parsons, J. H.; Saville-Stones, E. A. Fungicides. U.S. Patent US6432964B1, Aug 13, 2002. |
| 6. | Barbosa, J.; Carson, K. G.; Gardyan, M. W.; He, W.; Lombardo, V.; Pabba, P.; Tarver, J., Jr. Inhibitors of Notum pectinacylesterase and methods of their use. U.S. Pat. Appl. US 2012/0065200 A1, March 15, 2012. |
| 16. | Gillis, E. P.; Burke, M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204 |
| 13. | Atkinson, B. N.; Steadman, D.; Zhao, Y.; Sipthorp, J.; Vecchia, L.; Ruza, R. R.; Jeganathan, F.; Lines, G.; Frew, S.; Monaghan, A.; Kjær, S.; Bictash, M.; Jones, E. Y.; Fish, P. V. Med. Chem. Commun. 2019, 10, 1361–1369. doi:10.1039/c9md00096h |
| 8. | Manallack, D. T. Perspect. Med. Chem. 2007, 1, 25–38. doi:10.1177/1177391x0700100003 |
| 9. | Wager, T. T.; Chandrasekaran, R. Y.; Hou, X.; Troutman, M. D.; Verhoest, P. R.; Villalobos, A.; Will, Y. ACS Chem. Neurosci. 2010, 1, 420–434. doi:10.1021/cn100007x |
| 10. | Manallack, D. T.; Prankerd, R. J.; Yuriev, E.; Oprea, T. I.; Chalmers, D. K. Chem. Soc. Rev. 2013, 42, 485–496. doi:10.1039/c2cs35348b |
| 11. | Charifson, P. S.; Walters, W. P. J. Med. Chem. 2014, 57, 9701–9717. doi:10.1021/jm501000a |
| 12. | Di, L.; Rong, H.; Feng, B. J. Med. Chem. 2013, 56, 2–12. doi:10.1021/jm301297f |
| 14. | Mitchell, I. S.; Spencer, K. L.; Stengel, P.; Han, Y.; Kallan, N. C.; Munson, M.; Vigers, G. P. A.; Blake, J.; Piscopio, A.; Josey, J.; Miller, S.; Xiao, D.; Xu, R.; Rao, C.; Wang, B.; Bernacki, A. L. Akt protein kinase inhibitors. WO Patent WO2005/051304, June 9, 2005. |
© 2019 Willis et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)