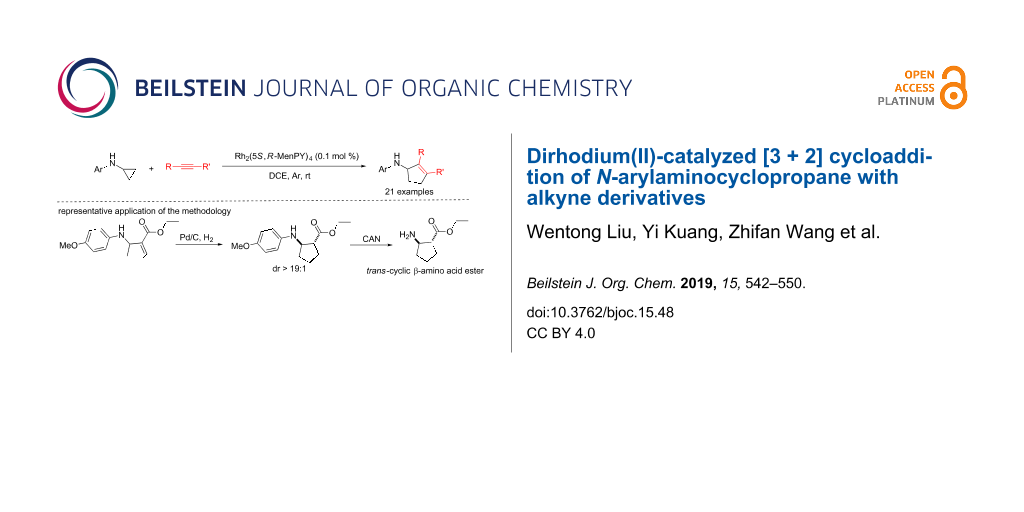
Abstract
Dirhodium(II) complex-catalyzed [3 + 2] reactions between N-arylaminocyclopropanes and alkyne derivatives are described. The cycloaddition products proved to be versatile synthetic intermediates. trans-Cyclic β-amino acids and derivatives thereof can be conveniently synthesized using this cycloaddition protocol.

Graphical Abstract
Introduction
N-Arylaminocyclopropanes 1 are important structural motifs for pharmaceuticals and are found especially in marketed fluoroquinolone antibiotics [1], such as ciprofloxacin and moxifloxacin (Scheme 1), and reverse transcriptase inhibitors [2], such as nevirapine. Meanwhile, since 1 contains a three-membered ring with high tension [3-6] and a nitrogen prone to single-electron oxidation, ring opening readily occurs followed by N-centered radical formation. The generated distonic radical cation can be further trapped by an alkene, alkyne, or triplet oxygen to initiate radical cyclization (Scheme 1) [7-15]. Thus, as key synthons, this class of molecules may play an important role in organic synthesis during construction of a series of aminocyclic compounds. In fact, the synthesis of 1 has always been a challenge [16,17]. Only recently an efficient synthesis method by arylation of cyclopropylamine has been developed and documented by Colacot et al. [16] and Stradiotto et al. [17], which has provided opportunities for further development of cycloaddition chemistry based on compound 1. Zheng et al. [7] first reported on the [3 + 2] cycloaddition reaction of 1 with an alkene or alkyne mediated by visible light by the aid of the photocatalyst [Ru(bpz)3](PF6)2. Our group reported the metal catalyst itself, particularly the dinuclear rhodium complex (Rh2(II,II)), that efficiently catalyzes the ring opening of 1 to achieve cycloaddition with alkene substrates under an argon atmosphere [18]. During the reaction, no metal valence changes were observed. We proposed that Rh2(II,II) may coordinate with the nitrogen in 1 to decrease the bond dissociation energy of N–H bonds, which may be beneficial to N-centered radical formation. However, due to the characteristics of the radical reaction, the resulting cycloaddition product mixtures have a low diastereoisomeric ratio, which increases the difficulty of separation and limits applications. In view of this, we applied alkyne derivatives as cycloaddition partners to make the developed methodology more applicable, as well as to investigate the possibility of chiral ring construction.
![[1860-5397-15-48-i1]](/bjoc/content/inline/1860-5397-15-48-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Applications of N-arylaminocyclopropanes.
Scheme 1: Applications of N-arylaminocyclopropanes.
Results and Discussion
Initially, phenylcyclopropylamine (1a) and ethyl propiolate (2a) were selected as substrates to study the cycloaddition in dichloromethane (DCM) with 1 mol % starting catalyst loading under the previously reported conditions [18]. In the absence of a catalyst, no cycloaddition product 3a formed (Table 1, entry 1). We then studied the effect of common rhodium catalysts on the reactions. When 1 mol % Rh(I) catalyst [Rh(CH2CH2)2Cl]2 was used (Table 1, entry 2), product 3a was not detected. The commonly used Rh(III) catalyst [Rh(Cp*)Cl2]2 lead to a 72% yield of product 3a (Table 1, entry 3). Next, the representative dirhodium(II) carboxylate such as Rh2(OAc)4, Rh2(TFA)4, Rh2(esp)2 and dirhodium(II) carboxamidate catalysts such as Rh2(cap)4, Rh2(5S, R-MenPY)4 were evaluated in the reaction. Rh2(OAc)4 created 3a with a yield of 45% (Table 1, entry 4) and Rh2(TFA)4 improved the yield of 3a to 52% (Table 1, entry 5). With the chelating catalyst Rh2(esp)2, the yield further increased to 68% (Table 1, entry 6). Although the carboxamidate type Rh2(cap)4 resulted in a lower yield (Table 1, entry 7), the yield raised to 61% when Rh2(5S, R-MenPY)4 [19,20] was applied as the catalyst (Table 1, entry 8). Next, 0.1 mol % catalytic loading was tested in the reactions to investigate the efficiency of the catalysts. Compared to the 1 mol % catalyst loading, the yield of 3a dropped to 39 and 44% when Rh2(esp)2 and [Rh(Cp*)Cl2]2, respectively, were used (Table 1, entries 9, 10), and kept practically consistent with Rh2(5S, R-MenPY)4 (Table 1, entry 11). These results indicate that the catalytic efficiency of Rh2(5S, R-MenPY)4 was the best of all the screened catalysts. Further solvent screening studies found the yields of 3a obtained in non-coordinating solvents, such as DCM, hexane, toluene, and 1,2-dichloroethane (DCE), and weak coordinating solvents, such as 1,2-dimethoxyethane (DME), were similar (Table 1, entries 11–15). DCE was the best solvent, leading to a 67% yield of 3a. Due to axial coordination of dirhodium(II) for a strong coordination solvent, the yield of the resulting cycloaddition product in N,N-dimethylformamide decreased to only 33% (Table 1, entry 16). Regrettably, though Rh2(5S,R-MenPY)4 is a chiral catalyst, the obtained cycloaddition products are racemic.
Table 1: Catalyst screening and optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-48-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Conditions | Solvent | Yieldb (%) |
| 1 | no catalyst | DCM | NR |
| 2 | [Rh(CH2CH2)2Cl]2 | DCM | ND |
| 3 | [Rh(Cp*)Cl2]2 | DCM | 72 |
| 4 | Rh2(OAc)4 | DCM | 45 |
| 5 | Rh2(TFA)4 | DCM | 52 |
| 6 | Rh2(esp)2 | DCM | 68 |
| 7 | Rh2(cap)4 | DCM | 40 |
| 8 | Rh2(5S,R-MenPY)4 | DCM | 61 |
| 9c | Rh2(esp)2 | DCM | 39 |
| 10c | [Rh(Cp*)Cl2]2 | DCM | 44 |
| 11c | Rh2(5S,R-MenPY)4 | DCM | 58 |
| 12c | Rh2(5S,R-MenPY)4 | DCE | 67 |
| 13c | Rh2(5S,R-MenPY)4 | hexane | 59 |
| 14c | Rh2(5S,R-MenPY)4 | toluene | 59 |
| 15c | Rh2(5S,R-MenPY)4 | DME | 60 |
| 16c | Rh2(5S,R-MenPY)4 | DMF | 33 |
aReaction conditions: 1a (0.5 mmol, 0.2 M in degassed solvent), 2a (2.5 mmol), catalyst (1 mol %) under argon at room temperature for 24 h unless otherwise noted. bIsolated yield. c0.1 mol % of catalyst. esp = α,α,α’,α’-tetramethyl-1,3-benzenedipropionate, cap = caprolactamate, 5S,R-MenPY = (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 2-oxopyrrolidine-5-carboxylate, NR = no reaction, ND = not detected.
According to previous reports [16], after a series of arylcyclopropylamines 1 with different substituents were synthesized, the scope of 1 was then explored and the results are shown in Table 2. The data suggest that the electronic effect on the aromatic ring of 1 influences the results of the reactions. Compound 1 with electron-donating groups, such as methoxy, tert-butyl, and methyl groups, all reacted smoothly to produce products with good yields (Table 2, entries 1–3). The cyclization of substrate 1 containing electron-withdrawing groups at the aromatic substuituent proceeded slowly, producing considerably lower yields of the corresponding products. For instance, multiple-substituted compound 1 (with a 3,5-disubstitued trifluoromethyl 1f) generated product 3f with a yield of 36% (88% brsm) after 24 h (Table 2, entry 5), but 3,5-dimethyl compound 1d afforded the desired product 3d with an excellent yield of 91% (Table 2, entry 3). We reasoned the electron-withdrawing groups on the arene increased the nitrogen–hydrogen bond dissociation energy (BDEN–H) of compound 1 [21], decreasing the rate of ring opening. Substrates 1i,j containing hindered substituents lead to low conversion of the substrates, producing only poor yields of products 3i,j (Table 2, entries 8 and 9), which implied that steric hindrance greatly influenced the reactions.
Table 2: Substrate scope of N-arylaminocyclopropanesa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-48-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yieldb | ||
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-48-i6.svg?max-width=637&scale=1.0)
|
1b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-48-i7.svg?max-width=637&scale=1.0)
|
3b | 59% |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-48-i8.svg?max-width=637&scale=1.0)
|
1c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-48-i9.svg?max-width=637&scale=1.0)
|
3c | 78% |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-48-i10.svg?max-width=637&scale=1.0)
|
1d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-48-i11.svg?max-width=637&scale=1.0)
|
3d | 91% |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-48-i12.svg?max-width=637&scale=1.0)
|
1e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-48-i13.svg?max-width=637&scale=1.0)
|
3e | 85% |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-48-i14.svg?max-width=637&scale=1.0)
|
1f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-48-i15.svg?max-width=637&scale=1.0)
|
3f | 36% (88% brsm) |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-48-i16.svg?max-width=637&scale=1.0)
|
1g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-48-i17.svg?max-width=637&scale=1.0)
|
3g | 67% |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-48-i18.svg?max-width=637&scale=1.0)
|
1h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-48-i19.svg?max-width=637&scale=1.0)
|
3h | 24% (52% brsm) |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-48-i20.svg?max-width=637&scale=1.0)
|
1i |
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-48-i21.svg?max-width=637&scale=1.0)
|
3i | 20% (67% brsm) |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-48-i22.svg?max-width=637&scale=1.0)
|
1j |
![[Graphic 20]](/bjoc/content/inline/1860-5397-15-48-i23.svg?max-width=637&scale=1.0)
|
3j | 37% (65% brsm) |
aReaction conditions: 1b–j (1 mmol, 0.2 M in degassed solvent), 2a (5 mmol), catalyst (0.1 mol %) at room temperature for 24 h unless otherwise noted. bIsolated yield; brsm = based on recovered starting material.
Next, we surveyed the different alkyne substrates 2 for cycloaddition under optimized reaction conditions. The terminal alkynes with electron-withdrawing groups reacted smoothly to produce the desired products, while alkyl-substituted terminal aldehydes, such as pent-1-yne (2b, Table 3, entry 1), did not produce the cycloaddition product [22-24]. tert-Butyldimethylsilyl-protected propargyl alcohol 2c had a greatly reduced reactivity (Table 3, entry 2) and the obtained yield was less than 10%. For aromatic terminal alkynes, moderate yields (Table 3, entries 3–6) were obtained regardless of the electron-donating or electron-withdrawing groups, indicating the electronic effect on the aromatic ring of 2 had little effect on the results of the reactions. Due to the decrease in the activity of the alkyne, the conjugated 2,4-hexadiyne (2h) produced a poor yield of only 31% (Table 3, entry 7). To study the chemical selectivity of the reaction, we examined conjugated alkyne substrates. For 2-methylbut-1-en-3-yne (2i) containing both terminal alkenes and alkynes, the activity of the olefin was greater than that of the alkyne, which favors alkene cycloaddition products with a ratio of 89:11. The observed selectivity of product 4i is similar to that previously reported [11] (Table 3, entry 8), indicating the results of this type of reaction are mainly determined by the stability of the free radical intermediate during the reaction. Further investigation revealed cycloaddition preferentially proceeded with the terminal alkene or alkyne. Product 4j was obtained with a 41% yield due to cycloaddition with an olefin group in 2j (Table 3, entry 9). When 2k was applied in the reaction, the addition product was obtained with a yield of 44% with the alkyne group being involved in the reaction (Table 3, entry 10). The reaction with 2l mainly produced 1,3-diene product 4l with a yield of 40% (Table 3, entry 11). In addition, we also examined some alkynes containing heterocycles, such as 3-ethynylpyridine and 2-ethynylthiophene. Because the heteroatoms were axially tightly coordinated to the dirhodium(II) catalyst, coordination of the cyclopropylamine with the dirhodium(II) catalyst was inhibited, resulting in no cycloaddition reaction occurring.
Table 3: Substrate scope of alkyne derivativesa.
![[Graphic 21]](/bjoc/content/inline/1860-5397-15-48-i24.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yieldb(%) | ||
| 1 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-15-48-i25.svg?max-width=637&scale=1.0)
|
2b | N.D. | 4b | N.R. |
| 2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-15-48-i26.svg?max-width=637&scale=1.0)
|
2c |
![[Graphic 24]](/bjoc/content/inline/1860-5397-15-48-i27.svg?max-width=637&scale=1.0)
|
4c | <10 |
| 3 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-15-48-i28.svg?max-width=637&scale=1.0)
|
2d |
![[Graphic 26]](/bjoc/content/inline/1860-5397-15-48-i29.svg?max-width=637&scale=1.0)
|
4d | 65 |
| 4 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-15-48-i30.svg?max-width=637&scale=1.0)
|
2e |
![[Graphic 28]](/bjoc/content/inline/1860-5397-15-48-i31.svg?max-width=637&scale=1.0)
|
4e | 64 |
| 5 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-15-48-i32.svg?max-width=637&scale=1.0)
|
2f |
![[Graphic 30]](/bjoc/content/inline/1860-5397-15-48-i33.svg?max-width=637&scale=1.0)
|
4f | 65 |
| 6 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-15-48-i34.svg?max-width=637&scale=1.0)
|
2g |
![[Graphic 32]](/bjoc/content/inline/1860-5397-15-48-i35.svg?max-width=637&scale=1.0)
|
4g | 64 |
| 7 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-15-48-i36.svg?max-width=637&scale=1.0)
|
2h |
![[Graphic 34]](/bjoc/content/inline/1860-5397-15-48-i37.svg?max-width=637&scale=1.0)
|
4h | 31 |
| 8c |
![[Graphic 35]](/bjoc/content/inline/1860-5397-15-48-i38.svg?max-width=637&scale=1.0)
|
2i |
![[Graphic 36]](/bjoc/content/inline/1860-5397-15-48-i39.svg?max-width=637&scale=1.0)
|
4id,e | 45 (17:83)f |
| 9 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-15-48-i40.svg?max-width=637&scale=1.0)
|
2j |
![[Graphic 38]](/bjoc/content/inline/1860-5397-15-48-i41.svg?max-width=637&scale=1.0) , ,
![[Graphic 39]](/bjoc/content/inline/1860-5397-15-48-i42.svg?max-width=637&scale=1.0)
|
4j | 41 (30:70)f |
| 10 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-15-48-i43.svg?max-width=637&scale=1.0)
|
2k |
![[Graphic 41]](/bjoc/content/inline/1860-5397-15-48-i44.svg?max-width=637&scale=1.0)
|
4k | 44 |
| 11c |
![[Graphic 42]](/bjoc/content/inline/1860-5397-15-48-i45.svg?max-width=637&scale=1.0)
|
2l |
![[Graphic 43]](/bjoc/content/inline/1860-5397-15-48-i46.svg?max-width=637&scale=1.0)
|
4ld | 40 |
aReaction conditions: 1a (1 mmol, 0.2 M in degassed solvent), 2b–l (5 mmol), catalyst (0.1 mol %) at room temperature for 24 h unless otherwise noted. bIsolated yield. cN-Arylaminocyclopropane 1d was used instead of 1a. dMajor isomer shown. eIsomer ratio 89:11. fDiastereoisomeric ratios (cis/trans) were determined by 1H NMR spectroscopy of the crude products.
To explore the synthetic practicality of this transformation, racemic compound 3b resulting from 1b and 2a was reduced using hydrogen and palladium on carbon (Scheme 2). It is interesting to note that only trans-5a was obtained and no cis product formed. After further removal of the para-methoxyphenyl (PMP) group using ammonium cerium nitrate (CAN), the cyclic β-amino acid ester 5b was obtained with a yield of 80%. Cyclic β-amino acids and derivatives have good bioactivity and are widely used as key synthetic intermediates in biomedical research [25-29]. Thus, based on this cycloaddition protocol, a convenient strategy can be established to synthesize trans-cyclic β-amino acids.
![[1860-5397-15-48-i2]](/bjoc/content/inline/1860-5397-15-48-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of trans-ethyl 2-aminocyclopentanecarboxylate.
Scheme 2: Synthesis of trans-ethyl 2-aminocyclopentanecarboxylate.
The mechanism of the [3 + 2] cycloaddition reaction of 1a and 2a is similar to that previously reported [18] (Scheme 3). The distonic radical cation C resulting from cyclopropane ring opening reacts with alkyne substrate 2a generating radical D. The intermediate radical D yielded E through intramolecular radical addition. After hydrogen atom transfer (HAT) from complex A, the desired product is obtained with regeneration of the N-centered radical B, which continues to catalyzing the reaction.
![[1860-5397-15-48-i3]](/bjoc/content/inline/1860-5397-15-48-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we report on the [3 + 2] cycloaddition reaction catalyzed by dirhodium(II) based on arylcyclopropylamine, which broadens the scope of this method to the alkynyl group. This study demonstrated that this cycloaddition method has potential synthetic practicality by providing a convenient way to synthesize trans-cyclic β-amino acid derivatives. Further application of this method with other cycloaddition partners and asymmetric synthesis of the chiral ring with the help of chiral auxiliaries are currently underway.
Experimental
General procedure for the [3 + 2] annulation of cyclopropylanilines: An oven-dried Schlenk tube equipped with a stirring bar was charged with Rh2(5S,R-MenPY)4 (0.1 mol %), alkyne (5.0 mmol), and dry DCE (5 mL). The tube was degassed through three freeze–pump–thaw cycles. After evacuating and backfilling the tube with argon three times, cyclopropylamine (1 mmol) was added. The reaction mixture was stirred at room temperature for 24 hours. After the reaction was complete, the mixture was concentrated and the residue was purified by flash chromatography to obtain the desired allylic amine.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization, and NMR spectra. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Garg, R.; Gupta, S. P.; Gao, H.; Babu, M. S.; Debnath, A. K.; Hansch, C. Chem. Rev. 1999, 99, 3525–3602. doi:10.1021/cr9703358
Return to citation in text: [1] -
Jiao, L.; Yu, Z.-X. J. Org. Chem. 2013, 78, 6842–6848. doi:10.1021/jo400609w
Return to citation in text: [1] -
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886
Return to citation in text: [1] -
Gao, Y.; Fu, X.-F.; Yu, Z.-X. Top. Curr. Chem. 2014, 346, 195–231. doi:10.1007/128_2014_527
Return to citation in text: [1] -
Grover, H. K.; Emmett, M. R.; Kerr, M. A. Org. Biomol. Chem. 2015, 13, 655–671. doi:10.1039/c4ob02117g
Return to citation in text: [1] -
Maity, S.; Zhu, M.; Shinabery, R. S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 222–226. doi:10.1002/anie.201106162
Return to citation in text: [1] [2] -
Maity, S.; Zheng, N. Synlett 2012, 23, 1851–1856. doi:10.1055/s-0032-1316592
Return to citation in text: [1] -
Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234
Return to citation in text: [1] -
Nguyen, T. H.; Maity, S.; Zheng, N. Beilstein J. Org. Chem. 2014, 10, 975–980. doi:10.3762/bjoc.10.96
Return to citation in text: [1] -
Nguyen, T. H.; Morris, S. A.; Zheng, N. Adv. Synth. Catal. 2014, 356, 2831–2837. doi:10.1002/adsc.201400742
Return to citation in text: [1] [2] -
Morris, S. A.; Wang, J.; Zheng, N. Acc. Chem. Res. 2016, 49, 1957–1968. doi:10.1021/acs.accounts.6b00263
Return to citation in text: [1] -
Cai, Y.; Wang, J.; Zhang, Y.; Li, Z.; Hu, D.; Zheng, N.; Chen, H. J. Am. Chem. Soc. 2017, 139, 12259–12266. doi:10.1021/jacs.7b06319
Return to citation in text: [1] -
Wimalasena, K.; Wickman, H. B.; Mahindaratne, M. P. D. Eur. J. Org. Chem. 2001, 3811–3817. doi:10.1002/1099-0690(200110)2001:20<3811::aid-ejoc3811>3.0.co;2-6
Return to citation in text: [1] -
Blackburn, A.; Bowles, D. M.; Curran, T. T.; Kim, H. Synth. Commun. 2012, 42, 1855–1863. doi:10.1080/00397911.2010.545166
Return to citation in text: [1] -
Gildner, P. G.; DeAngelis, A.; Colacot, T. J. Org. Lett. 2016, 18, 1442–1445. doi:10.1021/acs.orglett.6b00377
Return to citation in text: [1] [2] [3] -
Tassone, J. P.; MacQueen, P. M.; Lavoie, C. M.; Ferguson, M. J.; McDonald, R.; Stradiotto, M. ACS Catal. 2017, 7, 6048–6059. doi:10.1021/acscatal.7b02014
Return to citation in text: [1] [2] -
Kuang, Y.; Ning, Y.; Zhu, J.; Wang, Y. Org. Lett. 2018, 20, 2693–2697. doi:10.1021/acs.orglett.8b00904
Return to citation in text: [1] [2] [3] -
Wang, Y.; Wolf, J.; Zavalij, P.; Doyle, M. P. Angew. Chem., Int. Ed. 2008, 47, 1439–1442. doi:10.1002/anie.200704618
Return to citation in text: [1] -
Xu, X.; Wang, X.; Liu, Y.; Doyle, M. P. J. Org. Chem. 2014, 79, 12185–12190. doi:10.1021/jo5013674
Return to citation in text: [1] -
Lucarini, M.; Pedulli, G. F. Chem. Soc. Rev. 2010, 39, 2106–2119. doi:10.1039/b901838g
Return to citation in text: [1] -
Giese, B.; Lachhein, S. Angew. Chem., Int. Ed. Engl. 1982, 21, 768–775. doi:10.1002/anie.198207681
Return to citation in text: [1] -
Fischer, H.; Radom, L. Angew. Chem., Int. Ed. 2001, 40, 1340–1371. doi:10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-#
Return to citation in text: [1] -
Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d
Return to citation in text: [1] -
Gellman, S. H. Acc. Chem. Res. 1998, 31, 173–180. doi:10.1021/ar960298r
Return to citation in text: [1] -
Seebach, D.; Matthews, J. L. Chem. Commun. 1997, 2015–2022. doi:10.1039/a704933a
Return to citation in text: [1] -
Seebach, D.; Abele, S.; Gademann, K.; Guichard, G.; Hintermann, T.; Jaun, B.; Matthews, J. L.; Schreiber, J. V.; Oberer, L.; Hommel, U.; Widmer, H. Helv. Chim. Acta 1998, 81, 932–982. doi:10.1002/hlca.19980810513
Return to citation in text: [1] -
Davies, H. M. L.; Venkataramani, C. Angew. Chem., Int. Ed. 2002, 41, 2197–2199. doi:10.1002/1521-3773(20020617)41:12<2197::aid-anie2197>3.0.co;2-n
Return to citation in text: [1] -
Tang, W.; Wu, S.; Zhang, X. J. Am. Chem. Soc. 2003, 125, 9570–9571. doi:10.1021/ja035777h
Return to citation in text: [1]
| 18. | Kuang, Y.; Ning, Y.; Zhu, J.; Wang, Y. Org. Lett. 2018, 20, 2693–2697. doi:10.1021/acs.orglett.8b00904 |
| 1. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 16. | Gildner, P. G.; DeAngelis, A.; Colacot, T. J. Org. Lett. 2016, 18, 1442–1445. doi:10.1021/acs.orglett.6b00377 |
| 17. | Tassone, J. P.; MacQueen, P. M.; Lavoie, C. M.; Ferguson, M. J.; McDonald, R.; Stradiotto, M. ACS Catal. 2017, 7, 6048–6059. doi:10.1021/acscatal.7b02014 |
| 11. | Nguyen, T. H.; Morris, S. A.; Zheng, N. Adv. Synth. Catal. 2014, 356, 2831–2837. doi:10.1002/adsc.201400742 |
| 7. | Maity, S.; Zhu, M.; Shinabery, R. S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 222–226. doi:10.1002/anie.201106162 |
| 8. | Maity, S.; Zheng, N. Synlett 2012, 23, 1851–1856. doi:10.1055/s-0032-1316592 |
| 9. | Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234 |
| 10. | Nguyen, T. H.; Maity, S.; Zheng, N. Beilstein J. Org. Chem. 2014, 10, 975–980. doi:10.3762/bjoc.10.96 |
| 11. | Nguyen, T. H.; Morris, S. A.; Zheng, N. Adv. Synth. Catal. 2014, 356, 2831–2837. doi:10.1002/adsc.201400742 |
| 12. | Morris, S. A.; Wang, J.; Zheng, N. Acc. Chem. Res. 2016, 49, 1957–1968. doi:10.1021/acs.accounts.6b00263 |
| 13. | Cai, Y.; Wang, J.; Zhang, Y.; Li, Z.; Hu, D.; Zheng, N.; Chen, H. J. Am. Chem. Soc. 2017, 139, 12259–12266. doi:10.1021/jacs.7b06319 |
| 14. | Wimalasena, K.; Wickman, H. B.; Mahindaratne, M. P. D. Eur. J. Org. Chem. 2001, 3811–3817. doi:10.1002/1099-0690(200110)2001:20<3811::aid-ejoc3811>3.0.co;2-6 |
| 15. | Blackburn, A.; Bowles, D. M.; Curran, T. T.; Kim, H. Synth. Commun. 2012, 42, 1855–1863. doi:10.1080/00397911.2010.545166 |
| 25. | Gellman, S. H. Acc. Chem. Res. 1998, 31, 173–180. doi:10.1021/ar960298r |
| 26. | Seebach, D.; Matthews, J. L. Chem. Commun. 1997, 2015–2022. doi:10.1039/a704933a |
| 27. | Seebach, D.; Abele, S.; Gademann, K.; Guichard, G.; Hintermann, T.; Jaun, B.; Matthews, J. L.; Schreiber, J. V.; Oberer, L.; Hommel, U.; Widmer, H. Helv. Chim. Acta 1998, 81, 932–982. doi:10.1002/hlca.19980810513 |
| 28. | Davies, H. M. L.; Venkataramani, C. Angew. Chem., Int. Ed. 2002, 41, 2197–2199. doi:10.1002/1521-3773(20020617)41:12<2197::aid-anie2197>3.0.co;2-n |
| 29. | Tang, W.; Wu, S.; Zhang, X. J. Am. Chem. Soc. 2003, 125, 9570–9571. doi:10.1021/ja035777h |
| 3. | Jiao, L.; Yu, Z.-X. J. Org. Chem. 2013, 78, 6842–6848. doi:10.1021/jo400609w |
| 4. | Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886 |
| 5. | Gao, Y.; Fu, X.-F.; Yu, Z.-X. Top. Curr. Chem. 2014, 346, 195–231. doi:10.1007/128_2014_527 |
| 6. | Grover, H. K.; Emmett, M. R.; Kerr, M. A. Org. Biomol. Chem. 2015, 13, 655–671. doi:10.1039/c4ob02117g |
| 21. | Lucarini, M.; Pedulli, G. F. Chem. Soc. Rev. 2010, 39, 2106–2119. doi:10.1039/b901838g |
| 2. | Garg, R.; Gupta, S. P.; Gao, H.; Babu, M. S.; Debnath, A. K.; Hansch, C. Chem. Rev. 1999, 99, 3525–3602. doi:10.1021/cr9703358 |
| 22. | Giese, B.; Lachhein, S. Angew. Chem., Int. Ed. Engl. 1982, 21, 768–775. doi:10.1002/anie.198207681 |
| 23. | Fischer, H.; Radom, L. Angew. Chem., Int. Ed. 2001, 40, 1340–1371. doi:10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-# |
| 24. | Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d |
| 18. | Kuang, Y.; Ning, Y.; Zhu, J.; Wang, Y. Org. Lett. 2018, 20, 2693–2697. doi:10.1021/acs.orglett.8b00904 |
| 19. | Wang, Y.; Wolf, J.; Zavalij, P.; Doyle, M. P. Angew. Chem., Int. Ed. 2008, 47, 1439–1442. doi:10.1002/anie.200704618 |
| 20. | Xu, X.; Wang, X.; Liu, Y.; Doyle, M. P. J. Org. Chem. 2014, 79, 12185–12190. doi:10.1021/jo5013674 |
| 7. | Maity, S.; Zhu, M.; Shinabery, R. S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 222–226. doi:10.1002/anie.201106162 |
| 16. | Gildner, P. G.; DeAngelis, A.; Colacot, T. J. Org. Lett. 2016, 18, 1442–1445. doi:10.1021/acs.orglett.6b00377 |
| 17. | Tassone, J. P.; MacQueen, P. M.; Lavoie, C. M.; Ferguson, M. J.; McDonald, R.; Stradiotto, M. ACS Catal. 2017, 7, 6048–6059. doi:10.1021/acscatal.7b02014 |
| 16. | Gildner, P. G.; DeAngelis, A.; Colacot, T. J. Org. Lett. 2016, 18, 1442–1445. doi:10.1021/acs.orglett.6b00377 |
| 18. | Kuang, Y.; Ning, Y.; Zhu, J.; Wang, Y. Org. Lett. 2018, 20, 2693–2697. doi:10.1021/acs.orglett.8b00904 |
© 2019 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)