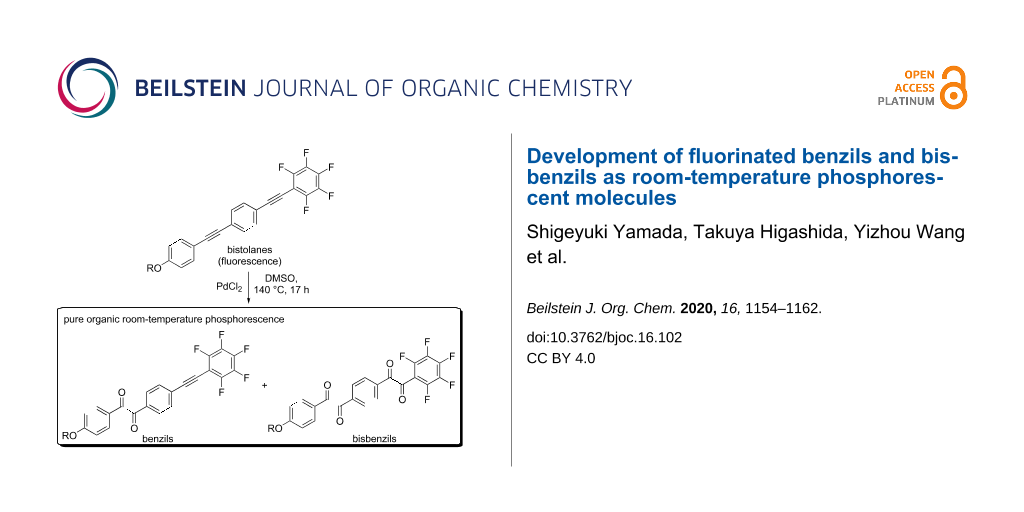
Abstract
Pure organic phosphorescent molecules are attractive alternatives to transition-metal-complex-based phosphores for biomedical and technological applications owing to their abundance and nontoxicity. This article discloses the design, synthesis, and photophysical properties of fluorinated benzil and bisbenzil derivatives as potential pure organic room-temperature phosphorescent molecules. These compounds were separately converted from the corresponding fluorinated bistolanes via PdCl2-catalyzed oxidation by dimethyl sulfoxide, while nonfluorinated bistolane provided the corresponding bisbenzil derivatives exclusively in a similar manner. Intensive investigations of the photophysical properties of the benzil and bisbenzil derivatives in toluene at 25 °C showed both fluorescence with a photoluminescence (PL) band at a maximum wavelength (λPL) of around 400 nm and phosphorescence with a PL band at a λPL of around 560 nm. Interestingly, intersystem crossing effectively caused fluorinated benzils to emit phosphorescence, which may arise from immediate spin-orbit coupling involving the 1(n, π)→3(π, π) transition, unlike the case of fluorinated or nonfluorinated bisbenzil analogues. These findings offer a useful guide for developing novel pure organic room-temperature phosphorescent materials.

Graphical Abstract
Introduction
The development of organic light-emitting molecules is recognized as one of the most important studies because of the broad application of these compounds as fluorescence probes, bio-imaging materials, and biosensors in biomedical diagnostics [1-4] and as organic light-emitting diodes in the technological field [5-8]. Among the organic light-emitting molecules developed thus far, extended π-conjugated compounds (e.g., pyrenes and perylenes) emit fluorescence, which is a radiative deactivation process from the lowest singlet (S1) excited state to the ground (S0) state [9]. Interestingly, for such π-conjugated molecules, suitable structural modifications can switch the radiative S1→S0 process to another radiative deactivation process from the triplet (T1) excited state to S0 via an S1→T1 intersystem crossing (ISC), resulting in the emission of phosphorescence [9].
Phosphorescent molecules generate two excitons (i.e., 25% S1 excitons and 75% T1 excitons) by application of an electric field, which is well known for organic light-emitting diodes. S1 excitons are converted to T1 excitons via an ISC process, finally achieving an excellent light-emitting efficiency (up to 100%) [10]. Therefore, extensive investigations to develop phosphorescent molecules have been performed thus far [11-13].
It has been established that transition metal complexes containing a heavy atom can promote ISC (e.g., Ru [14], Ir [15,16], Pt [17], and Au [18-21]) (Figure 1A), and this offers a molecular design approach for phosphorescence emission. However, it is becoming necessary to explore alternatives to rare metals because of the latter’s scarcity and toxicity. Owing to recent considerable efforts, several molecular designs have been proposed as alternatives, particularly the use of pure organic phosphorescent molecules [22].
![[1860-5397-16-102-1]](/bjoc/content/figures/1860-5397-16-102-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (A) Transition-metal-containing and (B) pure organic phosphorescent materials reported thus far (bpy: 2,2'-bipyridine, ppy: 2-phenylpyridine, OEP: octaethylporphyrin).
Figure 1: (A) Transition-metal-containing and (B) pure organic phosphorescent materials reported thus far (bp...
For example, methyl 4-bromobenzoate (Figure 1B) exhibits phosphorescence in the crystalline state via nonradiative ISC to the T1 state owing to crystallization-induced restriction of intramolecular motions [23]. Moreover, crystalline 2,5-dihexyloxy-4-bromobenzaldehyde displays green phosphorescence, which stems from rapid ISC due to the heavy atom effect via halogen bonding (C=O···Br) [24]. Moreover, benzophenone- or benzil-type molecules can achieve long-lived phosphorescence owing to a significant acceleration of spin-orbit coupling based on the El-Sayed rule involving the 1(n, π)→3(π, π) transition [25,26].
Over the past few years, our group has intensively investigated fluorinated 1,4-bis(2-phenylethyn-1-yl)benzenes (1), a structural class known as bistolanes (Figure 2A), which show prominent fluorescence not only in dilute solution, but also in the crystalline state [27-31].
![[1860-5397-16-102-2]](/bjoc/content/figures/1860-5397-16-102-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (A) Chemical structures of fluorescent bistolane derivatives previously developed by our group and (B) phosphorescent molecular structures intended for this work.
Figure 2: (A) Chemical structures of fluorescent bistolane derivatives previously developed by our group and ...
As a powerful tool to develop novel pure organic phosphorescent molecules, we envisioned the structural modification of the carbon–carbon triple (C≡C) bond in fluorinated bistolane 1 via oxidation to form the corresponding benzil 2 and/or bisbenzil 3 derivatives (Figure 2B). A literature review reveals that bisbenzil-type analogues have not received much attention, despite several publications on benzil-type phosphorescent molecules [25,26,32]. In this study, therefore, we examined the synthesis of novel benzil- and bisbenzil-type molecules via oxidation of fluorinated and nonfluorinated bistolane derivatives and evaluated their photophysical properties in detail.
Results and Discussion
Synthesis
This study was initiated with the synthesis of fluorinated bisbenzil derivatives. The PdCl2-catalyzed oxidation of the C≡C bonds in 1 by dimethyl sulfoxide (DMSO) was performed according to a previously reported procedure (Scheme 1) [33,34].
![[1860-5397-16-102-i1]](/bjoc/content/inline/1860-5397-16-102-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathway for fluorinated benzil (2) and bisbenzil (3) derivatives.
Scheme 1: Synthetic pathway for fluorinated benzil (2) and bisbenzil (3) derivatives.
The methoxy-substituted fluorinated bistolane 1a was prepared from commercially available 4-ethynylanisole in four facile steps. Interestingly, stirring 1a in DMSO solution in the presence of 30 mol % of PdCl2 at 140 °C for 17 h produced two products (56% yield for the more polar product and 37% yield for the less polar product) after purification with column chromatography. Spectroscopic analyses (i.e., 1H, 19F, and 13C nuclear magnetic resonance (NMR) spectroscopy, infrared spectroscopy, and high-resolution mass spectrometry) successfully identified the more polar product as the half-oxidized benzil 2a and the less polar one as the fully oxidized bisbenzil 3a. Fluorinated bistolane 1b, bearing a hexyloxy chain, also underwent PdCl2-catalyzed C≡C oxidation to give rise to the corresponding benzil 2b and bisbenzil 3b in 28% and 21% yield, respectively. When nonfluorinated bistolane 1c was used as the substrate, the corresponding bisbenzil 3c was obtained in 40% yield as the major product together with an inseparable mixture.
The proposed mechanism of Pd(II)-catalyzed C≡C oxidation is illustrated in Scheme 2 [33]: The catalytic cycle starts with the coordination of the electron-rich C≡C bond to the electron-deficient divalent Pd center, forming the corresponding π-complex (Int-A). Int-A smoothly undergoes nucleophilic attack by the oxygen atom in DMSO to construct a cationic vinylpalladium(II) species (Int-B). Further nucleophilic attack of another DMSO molecule against Int-B, followed by elimination of dimethyl sulfide, furnishes a cationic intermediate (Int-C). Finally, immediate elimination of dimethyl sulfide and the Pd(II) catalyst gives rise to the corresponding benzil 2, after which the eliminated Pd(II) catalyst is recycled to provide the oxidation products. The fully oxidized bisbenzil 3 is generated after further Pd(II)-catalyzed C≡C oxidation of the half-oxidized benzil 2 via the same catalytic cycle.
![[1860-5397-16-102-i2]](/bjoc/content/inline/1860-5397-16-102-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism of Pd(II)-catalyzed alkyne oxidation by dimethyl sulfoxide (DMSO).
Scheme 2: Proposed mechanism of Pd(II)-catalyzed alkyne oxidation by dimethyl sulfoxide (DMSO).
Considering the proposed reaction mechanism, the successful isolation of the half-oxidized benzil derivatives 2a and 2b from the oxidation of fluorinated bistolanes 1a and 1b, respectively, may be due to the decreased reactivity of the C≡C bond toward Pd(II)-catalyzed C≡C oxidation caused by the adjacent electron-deficient fluorinated aromatic ring. To confirm the electron-withdrawing effect of this fluorinated aromatic ring, the electronic charge at the adjacent C≡C bond was calculated by density functional theory (DFT) using the Gaussian 16 (Revision B.01) software package [35]. As typical examples, the molecular geometries of 1a and 1c were optimized at the CAM-B3LYP/6-31+G(d) level of theory. The absence of any imaginary frequency in the vibrational analysis proved that the calculated structures are the minima. Figure 3 shows the calculated Mulliken charge distributions of fluorinated 1a and nonfluorinated 1c.
![[1860-5397-16-102-3]](/bjoc/content/figures/1860-5397-16-102-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Mulliken charge distributions of fluorinated 1a and nonfluorinated 1c obtained from density functional theory calculations [CAM-B3LYP/6-31+G(d) level].
Figure 3: Mulliken charge distributions of fluorinated 1a and nonfluorinated 1c obtained from density functio...
The sp-hybridized carbon adjacent to the fluorinated aromatic ring of 1a has a significant positive Mulliken charge (+0.472), while that adjacent to the nonfluorinated aromatic ring of 1c has a negative charge (−0.304). This clearly indicates that the fluorinated aromatic ring retards the Pd(II)-catalyzed oxidation of the adjacent C≡C bond, thereby allowing the isolation of the half-oxidized benzil derivative 2a. On the basis of this theoretical investigation, the unique reactivities of bistolanes with fluorinated and nonfluorinated aromatic rings toward oxidation by DMSO can be rationalized.
Photophysical behavior
Our interest was then directed toward the photophysical properties of benzil and bisbenzil derivatives, which were freshly purified by column chromatography (eluent: hexane/EtOAc = 5:1 for benzil and 10:1 for bisbenzil) and subsequently recrystallized from hexane. The sample solution concentrations in toluene were 1.0 × 10−5 and 1.0 × 10−3 M for the absorption and photoluminescence (PL) measurements, respectively, and the absorption and PL spectra are shown in Figure 4. The photophysical data obtained from these measurements are summarized in Table 1.
![[1860-5397-16-102-4]](/bjoc/content/figures/1860-5397-16-102-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Absorption and photoluminescence (PL) spectra of (A) 2a, (B) 2b, (C) 3a, (D) 3b, and (E) 3c in toluene solution. Concentrations: 1.0 × 10−5 and 1.0 × 10−3 M for absorbance and PL measurements, respectively. Color legend: black: absorption, red: PL as prepared, green: PL under N2 atmosphere, and blue: PL under O2 atmosphere.
Figure 4: Absorption and photoluminescence (PL) spectra of (A) 2a, (B) 2b, (C) 3a, (D) 3b, and (E) 3c in tolu...
Table 1: Photophysical data from ultraviolet (UV)-visible absorption and steady-state photoluminescence (PL) measurementsa.
| λabs [nm] (ε [M−1 cm−1]) | λPL [nm]b (ΦPL)c | I560/I395 | |||
| pristine | N2 | O2 | |||
| 2a | 295 (28700), 315 (36500), 407 (160) |
395, 406 shd, 507, 563
(0.018) |
0.17 | 1.24 | 0.05 |
| 2b | 293 (40500), 314 (30400), 407 (180) |
395, 406 sh, 507, 563
(0.015) |
0.35 | 1.12 | 0.08 |
| 3a | 290 (30500), 405 (180) | 393, 406 sh, 516, 551 (<0.01) | 0.61 | 0.74 | 0.19 |
| 3b | 290 (51200), 405 (212) |
396, 412, 517, 554
(<0.01) |
0.79 | 0.84 | 0.50 |
| 3c | 290 (29600), 402 (260) |
397, 412, 514, 569
(<0.01) |
0.14 | 0.44 | 0.14 |
aToluene solution (concentrations: 1.0 × 10−5 and 1.0 × 10−3 M for UV-visible absorption and PL measurements, respectively); bExcitation wavelength: 350 nm; cQuantum yield measured using a calibrating sphere. Excitation wavelength: 290 nm. dShoulder peak.
The methoxy-substituted fluorinated benzil 2a in toluene absorbs UV light at 315 and 295 nm with molar extinction coefficients (ε) of 36500 and 28700 M−1·cm−1, respectively (Figure 4A). Similarly, the toluene solution of the fluorinated benzil with a hexyloxy chain (2b) absorbs UV light at 314 (ε: 30400 M−1·cm−1) and 293 nm (ε: 40500 M−1·cm−1) (Figure 4B). Both 2a and 2b exhibit weak absorption at around 400 nm (ε: ≈170 M−1·cm−1). As shown in Figure 4C–E, on the other hand, the bisbenzil derivatives 3a–c show an absorption band at 290 nm (ε: 29600–51200 M−1·cm−1) as the major signal, together with a quite weak absorption band at 402–405 nm (ε: 180–260 M−1·cm−1). To gain more information about the slight difference between the absorption behaviors of the benzil and bisbenzil derivatives, DFT and time-dependent DFT (TD-DFT) calculations at the CAM-B3LYP/6-31+G(d) level of theory were performed for fluorinated benzil 2a and bisbenzil derivative 3a as representative examples. Figure 5 shows the distributions of molecular orbitals involved in vertical electronic transitions in 2a and 3a.
![[1860-5397-16-102-5]](/bjoc/content/figures/1860-5397-16-102-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Distributions of molecular orbitals (isosurface value: 0.04 a.u.) involved in vertical electronic transitions in 2a and 3a calculated using density functional theory (DFT) and time-dependent DFT at the CAM-B3LYP/6-31+G(d) level (HOMO: highest occupied molecular orbital, LUMO: lowest unoccupied molecular orbital).
Figure 5: Distributions of molecular orbitals (isosurface value: 0.04 a.u.) involved in vertical electronic t...
The main electronic transition with a relatively large oscillator strength (f) in both 2a and 3a is the highest occupied molecular orbital (HOMO)→lowest unoccupied molecular orbital (LUMO) transition. Focusing on the orbital distribution, the HOMO lobe of 2a covers the entire molecule, while the LUMO lobe is localized at the tolane moiety. In the case of 3a, the HOMO lobe is localized at the methoxy-substituted benzene ring, while the LUMO lobe is localized at the central benzene ring. Accordingly, it can be concluded that the absorption bands at the short-wavelength region (around 290–315 nm) stems from the π–π* transition. The TD-DFT calculation also reveals that n–π* transitions (e.g., HOMO–2→LUMO for 2a and HOMO–3→LUMO for 3a) have small values of f; thus, the small absorption band at around 400 nm can be safely attributed to an n–π* transition.
Upon irradiation of the toluene solutions of benzil derivatives 2a and 2b with a 350 nm UV light, three PL bands at 395, 507, and 563 nm, along with a shoulder signal at around 406 nm, are observed (Figure 4A and 4B). The bisbenzil derivatives, i.e., 3a–c, also show similar PL behavior to the aforementioned benzil analogues: four PL bands with λPL of 393–397, 406–412, 514–517, and 551–569 nm are observed. To gain more information about the PL process in benzils and bisbenzils, the PL spectra of the toluene solutions (1.0 × 10−3 M) were acquired after bubbling with N2 or O2 gas for 30 min. In general, an O2-saturated environment strongly deactivates the triplet states; thus, PL emission stems only from fluorescence. On the other hand, elimination of O2 gas from a solution by bubbling with an inert gas (N2 or Ar) allows the triplet states to survive for a long lifetime, which possibly leads to a phosphorescence emission. Hence, the elimination of O2 gas from solutions by N2 gas bubbling (or addition of O2 gas in solutions) can judge the presence of phosphorescence, as well as the assignment of PL bands. The obtained PL spectra are superimposed on the PL spectra of a pristine sample (Figure 4). Upon bubbling the solution with N2 gas for 30 min, a dramatic enhancement of the PL intensities of benzils 2a and 2b at λPL = 563 nm is observed. The PL intensities of bisbenzils 3a–c at the long-wavelength region between 551 and 569 nm also increase, although the increment rates are not as high as those of 2a and 2b. On the contrary, bubbling the toluene solutions of benzil or bisbenzil derivatives with O2 gas causes the intensity of the long-wavelength PL band to decrease compared with that of the pristine solution, while the other remaining PL bands in the short-wavelength region do not change. Judging from the PL behavior under N2 or O2 flow conditions, the PL bands at the short-wavelength region around 395 nm and long-wavelength region around 560 nm can be safely considered fluorescence via radiative deactivation from the S1 excited state to the S0 state and phosphorescence via electronic transition from the T1 excited state to the S0 state, respectively. Accordingly, fluorinated benzils and bisbenzils show room-temperature phosphorescence in the solution state.
To understand the effects of structural modification (i.e., the benzil structure with a tolane vs bisbenzil moiety) and incorporation of fluorine atoms on the phosphorescence, the ratio between the peak intensities at ≈395 and ≈560 nm (I560/I395) was quantitatively calculated, and the results are summarized in Table 1. The I560/I395 values of benzils 2a and 2b under N2 flow conditions increase up to sevenfold compared with those of the corresponding pristine solutions. On the other hand, the increase in the I560/I395 values of fluorinated bisbenzil derivatives 3a and 3b is low (only 1.1 times) under N2 flow conditions, although the PL intensity of nonfluorinated 3c increases by approximately three times. Judging from these comprehensive observations, the benzil structure promotes ISC from S1 to T1, causing increment phosphorescence, unlike the corresponding bisbenzil scaffold. Moreover, fluorine substituents on the bisbenzil molecules cause significant retardation of ISC, leading to a weaker phosphorescence intensity.
Additionally, the quantum yields (ΦPL) of the PL bands in the range of 350–600 nm were acquired using an absolute quantum yield measurement system with a calibrated integrating sphere. All samples have a low ΦPL of less than 0.02 (Table 1), meaning that most of the excited states of all samples deactivate nonradiatively. At the moment, we cannot certify the origin of the low ΦPL in terms of the molecular properties, for instance, the main pathway of nonradiative deactivation. Experiments are currently being conducted to better understand the photophysical mechanisms of the excited-state dynamics of these benzil and bisbenzil derivatives.
Conclusion
In this article, we described the design and synthesis of benzil- or bisbenzil-based room-temperature phosphorescent molecules via a simple oxidation protocol for fluorescent bistolane derivatives. Nonfluorinated bistolane derivatives exclusively gave the corresponding products with a bisbenzil scaffold, whereas the fluorinated bistolane derivatives generated not only mono-oxidized benzil derivatives bearing a fluorinated tolane scaffold, but also the corresponding bis-oxidized bisbenzil derivatives. Based on theoretical calculations, the selective formation of the fluorinated analogues stemmed from the slight modulation of the charge distribution at the alkyne moiety of the reactant induced by the electron-withdrawing fluorine atoms. Evaluation of the photophysical behavior of the benzils and bisbenzils through several PL measurements under N2 and O2 flow conditions probed the successful room-temperature phosphorescence of the compounds in toluene solution. A fuller understanding of the excited-state dynamics of these benzil and bisbenzil derivatives will assist the development of environmentally benign, pure organic phosphorescent materials.
Supporting Information
Experimental procedures for the synthesis and characterization of fluorinated benzils 2a and 2b, fluorinated bisbenzils 3a and 3b, and nonfluorinated bisbenzil 3c. 1H, 13C, and 19F NMR spectra of 2a, 2b, and 3a–c. Cartesian coordinates of the optimized geometries of 1a, 1c, 2a, and 3a obtained from DFT calculations.
| Supporting Information File 1: Experimental preocedures, NMR spectra and Cartesian coordinates. | ||
| Format: PDF | Size: 2.3 MB | Download |
Funding
The following sources of funding are acknowledged: Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research (C) (Grant No. JP18K05262) and the AIST Nanocharacterization Facility (ANCF) platform as part of a program of the “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
-
Singh, H.; Tiwari, K.; Tiwari, R.; Pramanik, S. K.; Das, A. Chem. Rev. 2019, 119, 11718–11760. doi:10.1021/acs.chemrev.9b00379
Return to citation in text: [1] -
Mei, J.; Huang, Y.; Tian, H. ACS Appl. Mater. Interfaces 2018, 10, 12217–12261. doi:10.1021/acsami.7b14343
Return to citation in text: [1] -
Chen, X.; Wang, F.; Hyun, J. Y.; Wei, T.; Qiang, J.; Ren, X.; Shin, I.; Yoon, J. Chem. Soc. Rev. 2016, 45, 2976–3016. doi:10.1039/c6cs00192k
Return to citation in text: [1] -
Ma, Y.; Zhang, Y.; Yu, W. W. J. Mater. Chem. C 2019, 7, 13662–13679. doi:10.1039/c9tc04065j
Return to citation in text: [1] -
Yersin, H., Ed. Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence; Wiley-VCH: Weinheim, Germany, 2018. doi:10.1002/9783527691722
Return to citation in text: [1] -
Gaspar, D. J.; Polikarpov, E., Eds. OLED Fundamentals: Materials, Devices, and Processing of Organic Light-Emitting Diodes; CRC Press: Boca Raton, FL, USA, 2015. doi:10.1201/b18485
Return to citation in text: [1] -
Su, L.; Fan, X.; Yin, T.; Wang, H.; Li, Y.; Liu, F.; Li, J.; Zhang, H.; Xie, H. Adv. Opt. Mater. 2020, 8, 1900978. doi:10.1002/adom.201900978
Return to citation in text: [1] -
Ostroverkhova, O. Chem. Rev. 2016, 116, 13279–13412. doi:10.1021/acs.chemrev.6b00127
Return to citation in text: [1] -
Ronda, C. R. Emission and Excitation Mechanisms of Phosphors. In Luminescence: From Theory to Applications; Ronda, C. R., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp 1–34. doi:10.1002/9783527621064.ch1
Return to citation in text: [1] [2] -
Yersin, H.; Rausch, A. F.; Czerwieniec, R. Organometallic Emitters for OLEDs: Triplet Harvesting, Singlet Harvesting, Case Structures, and Trends. In Physics of Organic Semiconductors; Brütting, W.; Adachi, C., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 371–424. doi:10.1002/9783527654949.ch13
Return to citation in text: [1] -
Wang, W.; Zhang, Y.; Jin, W. J. Coord. Chem. Rev. 2020, 404, 213107. doi:10.1016/j.ccr.2019.213107
Return to citation in text: [1] -
Zhan, G.; Liu, Z.; Bian, Z.; Huang, C. Front. Chem. (Lausanne, Switz.) 2019, 7, 305. doi:10.3389/fchem.2019.00305
Return to citation in text: [1] -
Qu, G.; Zhang, Y.; Ma, X. Chin. Chem. Lett. 2019, 30, 1809–1814. doi:10.1016/j.cclet.2019.07.042
Return to citation in text: [1] -
Crosby, G. A. Acc. Chem. Res. 1975, 8, 231–238. doi:10.1021/ar50091a003
Return to citation in text: [1] -
Baldo, M. A.; Lamansky, S.; Burrows, P. E.; Thompson, M. E.; Forrest, S. R. Appl. Phys. Lett. 1999, 75, 4–6. doi:10.1063/1.124258
Return to citation in text: [1] -
Adachi, C.; Baldo, M. A.; Forrest, S. R.; Thompson, M. E. Appl. Phys. Lett. 2000, 77, 904–906. doi:10.1063/1.1306639
Return to citation in text: [1] -
Baldo, M. A.; O'Brien, D. F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M. E.; Forrest, S. R. Nature 1998, 395, 151–154. doi:10.1038/25954
Return to citation in text: [1] -
Yam, V. W.-W.; Wong, K. M.-C.; Hung, L.-L.; Zhu, N. Angew. Chem., Int. Ed. 2005, 44, 3107–3110. doi:10.1002/anie.200500253
Return to citation in text: [1] -
Yamada, S.; Yamaguchi, S.; Tsutsumi, O. J. Mater. Chem. C 2017, 5, 7977–7984. doi:10.1039/c7tc00728k
Return to citation in text: [1] -
Fujisawa, K.; Kawakami, N.; Onishi, Y.; Izumi, Y.; Tamai, S.; Sugimoto, N.; Tsutsumi, O. J. Mater. Chem. C 2013, 1, 5359–5366. doi:10.1039/c3tc31105h
Return to citation in text: [1] -
Seki, T.; Takamatsu, Y.; Ito, H. J. Am. Chem. Soc. 2016, 138, 6252–6260. doi:10.1021/jacs.6b02409
Return to citation in text: [1] -
Kenry; Chen, C.; Liu, B. Nat. Commun. 2019, 10, 2111. doi:10.1038/s41467-019-10033-2
Return to citation in text: [1] -
Yuan, W. Z.; Shen, X. Y.; Zhao, H.; Lam, J. W. Y.; Tang, L.; Lu, P.; Wang, C.; Liu, Y.; Wang, Z.; Zheng, Q.; Sun, J. Z.; Ma, Y.; Tang, B. Z. J. Phys. Chem. C 2010, 114, 6090–6099. doi:10.1021/jp909388y
Return to citation in text: [1] -
Bolton, O.; Lee, K.; Kim, H.-J.; Lin, K. Y.; Kim, J. Nat. Chem. 2011, 3, 205–210. doi:10.1038/nchem.984
Return to citation in text: [1] -
Zhao, W.; He, Z.; Lam, J. W. Y.; Peng, Q.; Ma, H.; Shuai, Z.; Bai, G.; Hao, J.; Tang, B. Z. Chem 2016, 1, 592–602. doi:10.1016/j.chempr.2016.08.010
Return to citation in text: [1] [2] -
Lower, S. K.; El-Sayed, M. A. Chem. Rev. 1966, 66, 199–241. doi:10.1021/cr60240a004
Return to citation in text: [1] [2] -
Morita, M.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Appl. Sci. 2019, 9, 1905. doi:10.3390/app9091905
Return to citation in text: [1] -
Yamada, S.; Miyano, K.; Agou, T.; Kubota, T.; Konno, T. Crystals 2019, 9, 195. doi:10.3390/cryst9040195
Return to citation in text: [1] -
Yamada, S.; Tanaka, T.; Ichikawa, T.; Konno, T. ACS Omega 2019, 4, 3922–3932. doi:10.1021/acsomega.8b03543
Return to citation in text: [1] -
Yamada, S.; Morita, M.; Agou, T.; Kubota, T.; Ichikawa, T.; Konno, T. Org. Biomol. Chem. 2018, 16, 5609–5617. doi:10.1039/c8ob01497c
Return to citation in text: [1] -
Yamada, S.; Miyano, K.; Konno, T.; Agou, T.; Kubota, T.; Hosokai, T. Org. Biomol. Chem. 2017, 15, 5949–5958. doi:10.1039/c7ob01369h
Return to citation in text: [1] -
Gong, Y. Y.; Tan, Y. Q.; Li, H.; Zhang, Y. R.; Yuan, W. Z.; Zhang, Y. M.; Sun, J. Z.; Tang, B. Z. Sci. China: Chem. 2013, 56, 1183–1186. doi:10.1007/s11426-013-4930-9
Return to citation in text: [1] -
Muzart, J. J. Mol. Catal. A: Chem. 2011, 338, 7–17. doi:10.1016/j.molcata.2011.01.030
Return to citation in text: [1] [2] -
Chi, K.-W.; Yusubov, M. S.; Filimonov, V. D. Synth. Commun. 1994, 24, 2119–2122. doi:10.1080/00397919408010224
Return to citation in text: [1] -
Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
Return to citation in text: [1]
| 33. | Muzart, J. J. Mol. Catal. A: Chem. 2011, 338, 7–17. doi:10.1016/j.molcata.2011.01.030 |
| 34. | Chi, K.-W.; Yusubov, M. S.; Filimonov, V. D. Synth. Commun. 1994, 24, 2119–2122. doi:10.1080/00397919408010224 |
| 33. | Muzart, J. J. Mol. Catal. A: Chem. 2011, 338, 7–17. doi:10.1016/j.molcata.2011.01.030 |
| 1. | Singh, H.; Tiwari, K.; Tiwari, R.; Pramanik, S. K.; Das, A. Chem. Rev. 2019, 119, 11718–11760. doi:10.1021/acs.chemrev.9b00379 |
| 2. | Mei, J.; Huang, Y.; Tian, H. ACS Appl. Mater. Interfaces 2018, 10, 12217–12261. doi:10.1021/acsami.7b14343 |
| 3. | Chen, X.; Wang, F.; Hyun, J. Y.; Wei, T.; Qiang, J.; Ren, X.; Shin, I.; Yoon, J. Chem. Soc. Rev. 2016, 45, 2976–3016. doi:10.1039/c6cs00192k |
| 4. | Ma, Y.; Zhang, Y.; Yu, W. W. J. Mater. Chem. C 2019, 7, 13662–13679. doi:10.1039/c9tc04065j |
| 10. | Yersin, H.; Rausch, A. F.; Czerwieniec, R. Organometallic Emitters for OLEDs: Triplet Harvesting, Singlet Harvesting, Case Structures, and Trends. In Physics of Organic Semiconductors; Brütting, W.; Adachi, C., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 371–424. doi:10.1002/9783527654949.ch13 |
| 27. | Morita, M.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Appl. Sci. 2019, 9, 1905. doi:10.3390/app9091905 |
| 28. | Yamada, S.; Miyano, K.; Agou, T.; Kubota, T.; Konno, T. Crystals 2019, 9, 195. doi:10.3390/cryst9040195 |
| 29. | Yamada, S.; Tanaka, T.; Ichikawa, T.; Konno, T. ACS Omega 2019, 4, 3922–3932. doi:10.1021/acsomega.8b03543 |
| 30. | Yamada, S.; Morita, M.; Agou, T.; Kubota, T.; Ichikawa, T.; Konno, T. Org. Biomol. Chem. 2018, 16, 5609–5617. doi:10.1039/c8ob01497c |
| 31. | Yamada, S.; Miyano, K.; Konno, T.; Agou, T.; Kubota, T.; Hosokai, T. Org. Biomol. Chem. 2017, 15, 5949–5958. doi:10.1039/c7ob01369h |
| 9. | Ronda, C. R. Emission and Excitation Mechanisms of Phosphors. In Luminescence: From Theory to Applications; Ronda, C. R., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp 1–34. doi:10.1002/9783527621064.ch1 |
| 25. | Zhao, W.; He, Z.; Lam, J. W. Y.; Peng, Q.; Ma, H.; Shuai, Z.; Bai, G.; Hao, J.; Tang, B. Z. Chem 2016, 1, 592–602. doi:10.1016/j.chempr.2016.08.010 |
| 26. | Lower, S. K.; El-Sayed, M. A. Chem. Rev. 1966, 66, 199–241. doi:10.1021/cr60240a004 |
| 32. | Gong, Y. Y.; Tan, Y. Q.; Li, H.; Zhang, Y. R.; Yuan, W. Z.; Zhang, Y. M.; Sun, J. Z.; Tang, B. Z. Sci. China: Chem. 2013, 56, 1183–1186. doi:10.1007/s11426-013-4930-9 |
| 9. | Ronda, C. R. Emission and Excitation Mechanisms of Phosphors. In Luminescence: From Theory to Applications; Ronda, C. R., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp 1–34. doi:10.1002/9783527621064.ch1 |
| 24. | Bolton, O.; Lee, K.; Kim, H.-J.; Lin, K. Y.; Kim, J. Nat. Chem. 2011, 3, 205–210. doi:10.1038/nchem.984 |
| 5. | Yersin, H., Ed. Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence; Wiley-VCH: Weinheim, Germany, 2018. doi:10.1002/9783527691722 |
| 6. | Gaspar, D. J.; Polikarpov, E., Eds. OLED Fundamentals: Materials, Devices, and Processing of Organic Light-Emitting Diodes; CRC Press: Boca Raton, FL, USA, 2015. doi:10.1201/b18485 |
| 7. | Su, L.; Fan, X.; Yin, T.; Wang, H.; Li, Y.; Liu, F.; Li, J.; Zhang, H.; Xie, H. Adv. Opt. Mater. 2020, 8, 1900978. doi:10.1002/adom.201900978 |
| 8. | Ostroverkhova, O. Chem. Rev. 2016, 116, 13279–13412. doi:10.1021/acs.chemrev.6b00127 |
| 25. | Zhao, W.; He, Z.; Lam, J. W. Y.; Peng, Q.; Ma, H.; Shuai, Z.; Bai, G.; Hao, J.; Tang, B. Z. Chem 2016, 1, 592–602. doi:10.1016/j.chempr.2016.08.010 |
| 26. | Lower, S. K.; El-Sayed, M. A. Chem. Rev. 1966, 66, 199–241. doi:10.1021/cr60240a004 |
| 17. | Baldo, M. A.; O'Brien, D. F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M. E.; Forrest, S. R. Nature 1998, 395, 151–154. doi:10.1038/25954 |
| 22. | Kenry; Chen, C.; Liu, B. Nat. Commun. 2019, 10, 2111. doi:10.1038/s41467-019-10033-2 |
| 15. | Baldo, M. A.; Lamansky, S.; Burrows, P. E.; Thompson, M. E.; Forrest, S. R. Appl. Phys. Lett. 1999, 75, 4–6. doi:10.1063/1.124258 |
| 16. | Adachi, C.; Baldo, M. A.; Forrest, S. R.; Thompson, M. E. Appl. Phys. Lett. 2000, 77, 904–906. doi:10.1063/1.1306639 |
| 23. | Yuan, W. Z.; Shen, X. Y.; Zhao, H.; Lam, J. W. Y.; Tang, L.; Lu, P.; Wang, C.; Liu, Y.; Wang, Z.; Zheng, Q.; Sun, J. Z.; Ma, Y.; Tang, B. Z. J. Phys. Chem. C 2010, 114, 6090–6099. doi:10.1021/jp909388y |
| 11. | Wang, W.; Zhang, Y.; Jin, W. J. Coord. Chem. Rev. 2020, 404, 213107. doi:10.1016/j.ccr.2019.213107 |
| 12. | Zhan, G.; Liu, Z.; Bian, Z.; Huang, C. Front. Chem. (Lausanne, Switz.) 2019, 7, 305. doi:10.3389/fchem.2019.00305 |
| 13. | Qu, G.; Zhang, Y.; Ma, X. Chin. Chem. Lett. 2019, 30, 1809–1814. doi:10.1016/j.cclet.2019.07.042 |
| 18. | Yam, V. W.-W.; Wong, K. M.-C.; Hung, L.-L.; Zhu, N. Angew. Chem., Int. Ed. 2005, 44, 3107–3110. doi:10.1002/anie.200500253 |
| 19. | Yamada, S.; Yamaguchi, S.; Tsutsumi, O. J. Mater. Chem. C 2017, 5, 7977–7984. doi:10.1039/c7tc00728k |
| 20. | Fujisawa, K.; Kawakami, N.; Onishi, Y.; Izumi, Y.; Tamai, S.; Sugimoto, N.; Tsutsumi, O. J. Mater. Chem. C 2013, 1, 5359–5366. doi:10.1039/c3tc31105h |
| 21. | Seki, T.; Takamatsu, Y.; Ito, H. J. Am. Chem. Soc. 2016, 138, 6252–6260. doi:10.1021/jacs.6b02409 |
© 2020 Yamada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)