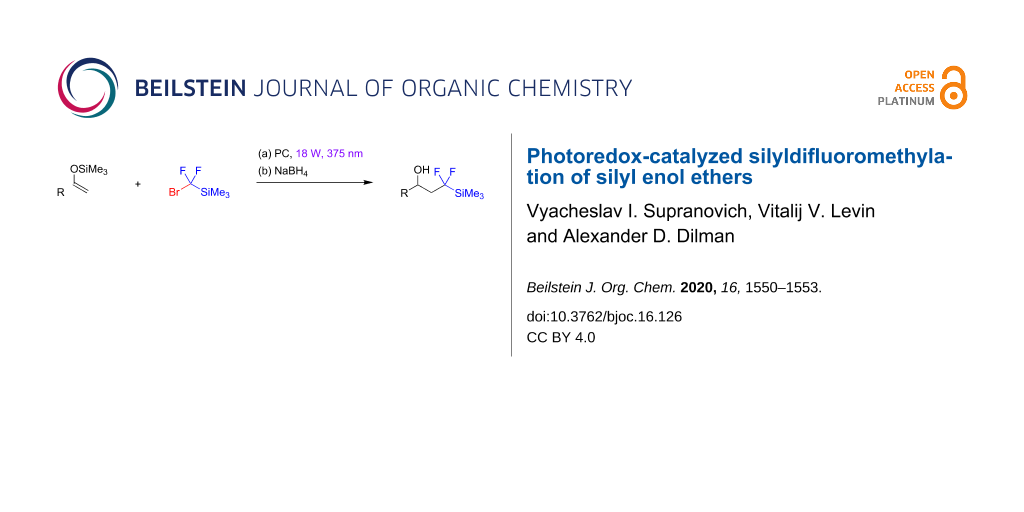
Abstract
A method for the light-mediated fluoroalkylation of silyl enol ethers with (bromodifluoromethyl)trimethylsilane followed by a reduction of the primary products with sodium borohydride is described. An 18 W, 375 nm LED was used as the light source. The reaction is performed in the presence of a gold photocatalyst, which effects the generation of a (trimethylsilyl)difluoromethyl radical via cleavage of the carbon–bromine bond.

Graphical Abstract
Findings
Fluorinated silicon reagents have found widespread use for the introduction of fluorinated fragments [1-5]. Typically, these reagents work under strongly basic conditions required to activate inert C–Si bonds with the generation of carbanionic species. On the other hand, radical reactions offer different synthetic opportunities originating from the neutral character of the intermediates [6,7] and, accordingly, radical fluoroalkylation processes have been extensively investigated over the last decade [8-13].
Recently, (bromodifluoromethyl)trimethylsilane (1) which can be readily obtained from the Ruppert–Prakash reagent [14,15], has been introduced as a reagent for the synthesis of various difluorinated compounds [16-18]. This silane is very sensitive to Lewis bases and accordingly it was used as a precursor of difluorocarbene, which can react with enol ethers [19,20] (Scheme 1). We showed that this silane could be involved in the radical chain hydrofluoroalkylation of electron-deficient alkenes, using a boron hydride as a source of hydrogen [21]. We thought that silane 1 could couple with silyl enol ethers in the presence of a photocatalyst affording fluoroalkylation products. Indeed, silyl enol ethers were found to be good acceptors of fluorinated radicals, and the resultant silyloxy-substituted radicals underwent single-electron oxidation thereby supporting a photoredox cycle [22-24].
![[1860-5397-16-126-i1]](/bjoc/content/inline/1860-5397-16-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of (bromodifluoromethyl)trimethylsilane (1).
Scheme 1: Reactions of (bromodifluoromethyl)trimethylsilane (1).
The silyl enol ether 2a derived from acetophenone was selected as a model substrate and the reaction with silane 1 (1.5 equiv) was evaluated (Scheme 2). The reactions were performed in dichloromethane, and reaction mixtures were analyzed by 19F NMR spectroscopy. A series of typical photocatalysts (for example, iridium catalysts) were ineffective in promoting the reaction. Rewardingly, a gold catalyst, [AuCl(μ-dppm)]2 [25-27], provided reasonable yields of 3a after one day of irradiation along with a full conversion of the starting enol ether. Moreover, a further increase in reaction time was accompanied by a decrease in the product yield. Finally, GC monitoring suggested complete consumption of the silyl enol ether within 6 hours. It should also be pointed out that the addition of basic additives, which are frequently employed in photoredox reactions to scavenge acidic byproducts [28,29], could not be employed. Silane 1 is easily destroyed by bases (even by the amide group [30]) followed by the rapid addition of difluorocarbene to silyl enol ethers [19,20]. Disappointingly, we were unable to isolate ketone 3a using flash column chromatography on silica gel, presumably, owing to facile β-elimination of hydrogen fluoride. To isolate a stable product, the reaction mixture was treated with sodium borohydride in ethanol, which effected the reduction of the keto group affording the corresponding alcohol 4a in 52% yield.
![[1860-5397-16-126-i2]](/bjoc/content/inline/1860-5397-16-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Optimization studies. Yield determined by 19F NMR spectroscopy using an internal standard.
Scheme 2: Optimization studies. Yield determined by 19F NMR spectroscopy using an internal standard.
Under the optimized conditions, a series of silyl enol ethers 2 were reacted with silane 1 (Figure 1). The reaction worked well with enol ethers derived from aromatic ketones, while those obtained from aliphatic ketones were ineffective. This may be ascribed to the decreased radical-stabilizing effect of the alkyl group compared to that of an aryl group, which either makes radical addition reversible or attenuates the reactivity of the starting enol ether. Similarly, an aromatic substrate bearing an ortho substituent gave lower yields, which may be associated with the ability of the ortho group to disfavor the planar conformation needed to stabilize an intermediate benzyl radical. The complete conversions of the enol ethers were usually achieved within 6 hours, though to obtain products 4f,k a longer time of 24 h was needed. Generally, higher yields were observed with substrates containing electron-donating functional groups. Substrates containing pyridine, furan, and thiophene as heterocyclic fragments were also successfully converted into the corresponding alcohols 4. However, in the reaction of the enol ether derived from 2-acetyl-N-methylpyrrole, ketone 3p did not undergo reduction with sodium borohydride, and the decreased reactivity of this ketone allowed its isolation.
![[1860-5397-16-126-1]](/bjoc/content/figures/1860-5397-16-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Reaction of silyl enol ethers. Yields refer to isolated yields. aReaction time 24 h; b1.0 equiv of silane 1 was used; cketone was isolated.
Figure 1: Reaction of silyl enol ethers. Yields refer to isolated yields. aReaction time 24 h; b1.0 equiv of ...
A proposed mechanism for the photoredox fluoroalkylation reaction is shown in Scheme 3. The photoexcited catalyst converts silane 1 into difluoromethylsilyl-based radical. The efficiency of [AuCl(μ-dppm)]2 compared to other strongly reducing catalysts may be associated with the ability of gold to interact with the bromine atom of silane 1 followed by inner-sphere electron transfer [27]. The radical then attacks silyl enol ether 2, and the subsequent silyloxy-substituted radical is oxidized by the photocatalyst to generate the product with concomitant production of TMSBr.
![[1860-5397-16-126-i3]](/bjoc/content/inline/1860-5397-16-126-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of the fluoroalkylation reaction.
Scheme 3: Proposed mechanism of the fluoroalkylation reaction.
Conclusion
In summary, a method for the introduction of the silyldifluoromethyl group was described by the coupling of a readily available silicon reagent with silyl enol ethers. The reaction is promoted by light and involves the generation of a fluorinated carbon-based radical via cleavage of the carbon–bromine bond by a gold photocatalyst.
Supporting Information
| Supporting Information File 1: Full experimental details, compound characterization, and copies of NMR spectra. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] -
Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a
Return to citation in text: [1] -
Krishnamoorthy, S.; Prakash, G. K. S. Synthesis 2017, 49, 3394–3406. doi:10.1055/s-0036-1588489
Return to citation in text: [1] -
Uneyama, K. J. Fluorine Chem. 2008, 129, 550–576. doi:10.1016/j.jfluchem.2008.05.012
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V. Mendeleev Commun. 2015, 25, 239–244. doi:10.1016/j.mencom.2015.07.001
Return to citation in text: [1] -
Yan, M.; Lo, J. C.; Edwards, J. T.; Baran, P. S. J. Am. Chem. Soc. 2016, 138, 12692–12714. doi:10.1021/jacs.6b08856
Return to citation in text: [1] -
Zard, S. Z. Org. Lett. 2017, 19, 1257–1269. doi:10.1021/acs.orglett.7b00531
Return to citation in text: [1] -
Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. doi:10.1002/anie.201202624
Return to citation in text: [1] -
Barata-Vallejo, S.; Cooke, M. V.; Postigo, A. ACS Catal. 2018, 8, 7287–7307. doi:10.1021/acscatal.8b02066
Return to citation in text: [1] -
Koike, T.; Akita, M. Acc. Chem. Res. 2016, 49, 1937–1945. doi:10.1021/acs.accounts.6b00268
Return to citation in text: [1] -
Koike, T.; Akita, M. Org. Biomol. Chem. 2019, 17, 5413–5419. doi:10.1039/c9ob00734b
Return to citation in text: [1] -
Lemos, A.; Lemaire, C.; Luxen, A. Adv. Synth. Catal. 2019, 361, 1500–1537. doi:10.1002/adsc.201801121
Return to citation in text: [1] -
Dagousset, G.; Carboni, A.; Masson, G.; Magnier, E. Visible Light–Induced (Per)fluoroalkylation by Photoredox Catalysis. In Modern Synthesis Processes and Reactivity of Fluorinated Compounds; Groult, H.; Leroux, F. R.; Tressaud, A., Eds.; Elsevier, 2017; pp 389–426. doi:10.1016/b978-0-12-803740-9.00014-7
Return to citation in text: [1] -
Kosobokov, M. D.; Dilman, A. D.; Levin, V. V.; Struchkova, M. I. J. Org. Chem. 2012, 77, 5850–5855. doi:10.1021/jo301094b
Return to citation in text: [1] -
Li, L.; Wang, F.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2013, 52, 12390–12394. doi:10.1002/anie.201306703
Return to citation in text: [1] -
Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 3206–3210. doi:10.1002/anie.201611823
Return to citation in text: [1] -
Xie, Q.; Zhu, Z.; Li, L.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2019, 58, 6405–6410. doi:10.1002/anie.201900763
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V. Acc. Chem. Res. 2018, 51, 1272–1280. doi:10.1021/acs.accounts.8b00079
Return to citation in text: [1] -
Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2015, 17, 760–763. doi:10.1021/acs.orglett.5b00097
Return to citation in text: [1] [2] -
Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904
Return to citation in text: [1] [2] -
Supranovich, V. I.; Levin, V. V.; Struchkova, M. I.; Korlyukov, A. A.; Dilman, A. D. Org. Lett. 2017, 19, 3215–3218. doi:10.1021/acs.orglett.7b01334
Return to citation in text: [1] -
Pham, P. V.; Nagib, D. A.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2011, 50, 6119–6122. doi:10.1002/anie.201101861
Return to citation in text: [1] -
Panferova, L. I.; Tsymbal, A. V.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2016, 18, 996–999. doi:10.1021/acs.orglett.6b00117
Return to citation in text: [1] -
Chernov, G. N.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. Adv. Synth. Catal. 2017, 359, 3063–3067. doi:10.1002/adsc.201700423
Return to citation in text: [1] -
Zidan, M.; Rohe, S.; McCallum, T.; Barriault, L. Catal. Sci. Technol. 2018, 8, 6019–6028. doi:10.1039/c8cy01765d
Return to citation in text: [1] -
Revol, G.; McCallum, T.; Morin, M.; Gagosz, F.; Barriault, L. Angew. Chem., Int. Ed. 2013, 52, 13342–13345. doi:10.1002/anie.201306727
Return to citation in text: [1] -
Li, D.; Che, C.-M.; Kwong, H.-L.; Yam, V. W.-W. J. Chem. Soc., Dalton Trans. 1992, 3325–3329. doi:10.1039/dt9920003325
Return to citation in text: [1] [2] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449
Return to citation in text: [1] -
Tsymbal, A. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2014, 79, 7831–7835. doi:10.1021/jo501644m
Return to citation in text: [1]
| 1. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 2. | Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a |
| 3. | Krishnamoorthy, S.; Prakash, G. K. S. Synthesis 2017, 49, 3394–3406. doi:10.1055/s-0036-1588489 |
| 4. | Uneyama, K. J. Fluorine Chem. 2008, 129, 550–576. doi:10.1016/j.jfluchem.2008.05.012 |
| 5. | Dilman, A. D.; Levin, V. V. Mendeleev Commun. 2015, 25, 239–244. doi:10.1016/j.mencom.2015.07.001 |
| 16. | Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 3206–3210. doi:10.1002/anie.201611823 |
| 17. | Xie, Q.; Zhu, Z.; Li, L.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2019, 58, 6405–6410. doi:10.1002/anie.201900763 |
| 18. | Dilman, A. D.; Levin, V. V. Acc. Chem. Res. 2018, 51, 1272–1280. doi:10.1021/acs.accounts.8b00079 |
| 14. | Kosobokov, M. D.; Dilman, A. D.; Levin, V. V.; Struchkova, M. I. J. Org. Chem. 2012, 77, 5850–5855. doi:10.1021/jo301094b |
| 15. | Li, L.; Wang, F.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2013, 52, 12390–12394. doi:10.1002/anie.201306703 |
| 8. | Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. doi:10.1002/anie.201202624 |
| 9. | Barata-Vallejo, S.; Cooke, M. V.; Postigo, A. ACS Catal. 2018, 8, 7287–7307. doi:10.1021/acscatal.8b02066 |
| 10. | Koike, T.; Akita, M. Acc. Chem. Res. 2016, 49, 1937–1945. doi:10.1021/acs.accounts.6b00268 |
| 11. | Koike, T.; Akita, M. Org. Biomol. Chem. 2019, 17, 5413–5419. doi:10.1039/c9ob00734b |
| 12. | Lemos, A.; Lemaire, C.; Luxen, A. Adv. Synth. Catal. 2019, 361, 1500–1537. doi:10.1002/adsc.201801121 |
| 13. | Dagousset, G.; Carboni, A.; Masson, G.; Magnier, E. Visible Light–Induced (Per)fluoroalkylation by Photoredox Catalysis. In Modern Synthesis Processes and Reactivity of Fluorinated Compounds; Groult, H.; Leroux, F. R.; Tressaud, A., Eds.; Elsevier, 2017; pp 389–426. doi:10.1016/b978-0-12-803740-9.00014-7 |
| 27. | Li, D.; Che, C.-M.; Kwong, H.-L.; Yam, V. W.-W. J. Chem. Soc., Dalton Trans. 1992, 3325–3329. doi:10.1039/dt9920003325 |
| 6. | Yan, M.; Lo, J. C.; Edwards, J. T.; Baran, P. S. J. Am. Chem. Soc. 2016, 138, 12692–12714. doi:10.1021/jacs.6b08856 |
| 7. | Zard, S. Z. Org. Lett. 2017, 19, 1257–1269. doi:10.1021/acs.orglett.7b00531 |
| 25. | Zidan, M.; Rohe, S.; McCallum, T.; Barriault, L. Catal. Sci. Technol. 2018, 8, 6019–6028. doi:10.1039/c8cy01765d |
| 26. | Revol, G.; McCallum, T.; Morin, M.; Gagosz, F.; Barriault, L. Angew. Chem., Int. Ed. 2013, 52, 13342–13345. doi:10.1002/anie.201306727 |
| 27. | Li, D.; Che, C.-M.; Kwong, H.-L.; Yam, V. W.-W. J. Chem. Soc., Dalton Trans. 1992, 3325–3329. doi:10.1039/dt9920003325 |
| 30. | Tsymbal, A. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2014, 79, 7831–7835. doi:10.1021/jo501644m |
| 22. | Pham, P. V.; Nagib, D. A.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2011, 50, 6119–6122. doi:10.1002/anie.201101861 |
| 23. | Panferova, L. I.; Tsymbal, A. V.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2016, 18, 996–999. doi:10.1021/acs.orglett.6b00117 |
| 24. | Chernov, G. N.; Levin, V. V.; Kokorekin, V. A.; Struchkova, M. I.; Dilman, A. D. Adv. Synth. Catal. 2017, 359, 3063–3067. doi:10.1002/adsc.201700423 |
| 19. | Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2015, 17, 760–763. doi:10.1021/acs.orglett.5b00097 |
| 20. | Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904 |
| 21. | Supranovich, V. I.; Levin, V. V.; Struchkova, M. I.; Korlyukov, A. A.; Dilman, A. D. Org. Lett. 2017, 19, 3215–3218. doi:10.1021/acs.orglett.7b01334 |
| 19. | Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2015, 17, 760–763. doi:10.1021/acs.orglett.5b00097 |
| 20. | Fedorov, O. V.; Kosobokov, M. D.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. J. Org. Chem. 2015, 80, 5870–5876. doi:10.1021/acs.joc.5b00904 |
| 28. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 29. | Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449 |
© 2020 Supranovich et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)