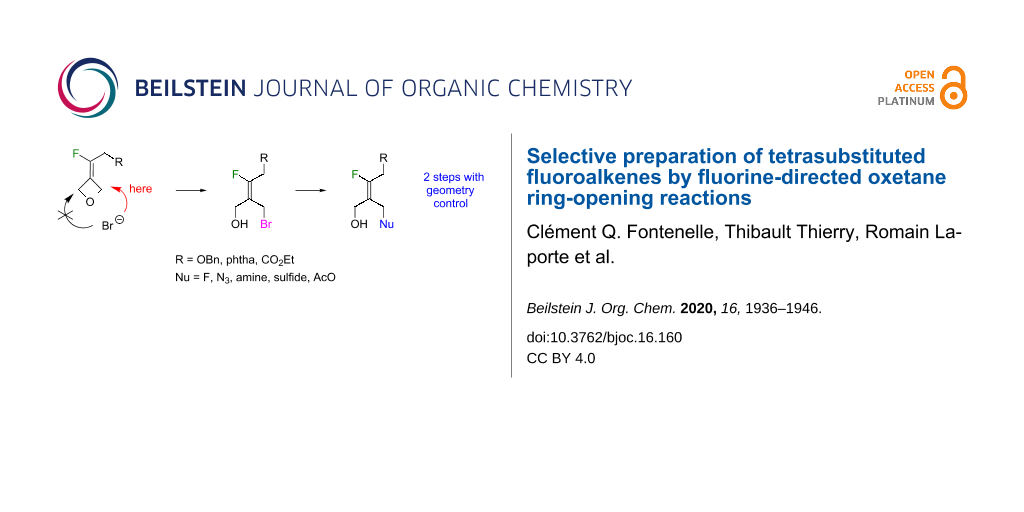
Abstract
The selective ring-opening reaction of fluoroalkylidene-oxetanes was directed by the presence of the fluorine atom, enabling a two-step access to tetrasubstituted fluoroalkenes with excellent geometry control. Despite its small van der Waals radii electronic, rather than steric influences of the fluorine atom governed the ring-opening reaction with bromide ions, even in the presence of bulky substituents.

Graphical Abstract
Introduction
The introduction of fluorine atoms into organic compounds is known to modify their biological and physiological properties and can enhance the half-life of drugs in vivo [1-4]. During the last decade, fluorinated nucleoside analogues have received increasing interest, as is illustrated by the two pharmaceutical leads gemcitabine (I) and sofosbuvir (II), potent anticancer or antiviral agents, respectively (Figure 1) [5,6]. The field of acyclonucleotides (ACN) has been explored less, however, the introduction of fluorine atoms showed remarkable effects. The most representative examples are phosphate analogues such as the nucleoside phosphorylase inhibitor III and acyclic nucleotides such as the antiviral agent FPMPA (IV) [7-9].
![[1860-5397-16-160-1]](/bjoc/content/figures/1860-5397-16-160-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative fluorinated nucleos(t)ides and acyclonucleotides.
Figure 1: Representative fluorinated nucleos(t)ides and acyclonucleotides.
Other main structural modifications of ACN relied on the introduction of a hydroxy group into the aliphatic chain to improve hydrogen bonding with enzymes [10], or of a carbon–carbon double bond to constrain the aliphatic chain and to limit conformational changes [11-13]. For the latter, nucleoside analogues (Figure 2, VI) containing a trans-butenyl moiety where the endocyclic C–O bond was replaced by a C=C bond are recognized by kinases as dUMP surrogate (V) [11]. However, there is no existing data for the corresponding fluoroalkene (VII), as the latter was not yet synthetized. It is expected that the introduction of fluorine into the carbon–carbon double bond, in a position equivalent to the ring oxygen of the naturally occurring nucleotide, will improve molecular recognition and activity. In addition, the polarity of the nucleotide and hydrogen-bond accepting capacity with proteins or enzymes would be restored [14].
![[1860-5397-16-160-2]](/bjoc/content/figures/1860-5397-16-160-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Acyclonucleotides as nucleotide surrogates.
Figure 2: Acyclonucleotides as nucleotide surrogates.
The synthesis of fluoroalkene precursors of modified acyclonucleosides (VIII) has been explored by Choi, and more recently by us [15-17]. Nevertheless, it was reported that no antiviral activity for compounds of series VIII was observed due to the difficulty of phosphorylation of the substrate by kinases [16]. The first kinase phosphorylation step is generally rate limiting, and the prior introduction of a phosphate or phosphonate function can circumvent this problem. The preparation of diols VIII was realized by olefination of a protected 1,3-dihydroxypropanone (Figure 3). However, the selective introduction of functional groups is not possible in these diols as the two hydroxy groups present similar chemical reactivity. Other approaches are available for a selective preparation of monofluoroalkenes including olefination or defluorination reactions or a sigmatropic rearrangement, but these approaches are limited and do not allow the synthesis of tetrasubstituted fluoroalkenes with good control of their geometry [18-21]. In order to develop a selective synthesis for tetrasubstituted fluoroalkenes we envisioned an alternative approach starting from fluoroalkylidene-oxetane derivatives and to the end we have studied the selectivity of the oxetane ring-opening reaction (Figure 3).
![[1860-5397-16-160-3]](/bjoc/content/figures/1860-5397-16-160-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Olefination approaches and ring-opening of oxetane derivatives.
Figure 3: Olefination approaches and ring-opening of oxetane derivatives.
Results and Discussion
The preparation of a series of fluoroalkylidene-oxetanes 1–3 was previously reported from 3-oxetanone through an olefination reaction with benzothiazoyl sulfones (Scheme 1) [22]. With these fluoroalkylidene-oxetanes in hands, we studied the selectivity of ring-opening reactions with heteroatom nucleophiles in order to access tetrasubstituted fluoroalkenes. A control of the geometry of these reactions would allow ready access to novel fluorinated ACN precursors.
![[1860-5397-16-160-i1]](/bjoc/content/inline/1860-5397-16-160-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of fluoroakylidene-oxetanes and their ring-opening reactions.
Scheme 1: Preparation of fluoroakylidene-oxetanes and their ring-opening reactions.
At the outset the opening of the oxetane ring of 1 by a range of nucleophiles was trialed under acidic conditions. Inspired by Yadav et al. [23], methanol (20 equiv) was used as nucleophile in the presence of camphorsulfonic acid (CSA, 1 equiv) in dichloromethane. The reaction was slow and required heating (50–65 °C) for 26 h to reach a 75% conversion and afforded a mixture of E/Z-1a in 60% yield. However, a low E/Z selectivity (40:60) was observed (Table 1, entry 1). When using neat benzylic alcohol, completion was achieved after 20 h at 80 °C but again no substantial selectivity could be observed for product 1b (Table 1, entry 2). Inspired by the work of Müller and Wang [24,25], substitution by the more nucleophilic 2-mercaptobenzothiazol (BTSH, 1.4 equiv) was also possible in the presence of CSA (1 equiv) at 20 °C and an 80% conversion was reached after 24 h. An improved E/Z ratio of 25:75 was determined for this reaction and compound 1c was isolated in 65% yield (Table 1, entry 3). In this case the nucleophilic ring-opening reaction appeared to be controlled by steric repulsions between the bulky benzothiazolyl and phthalimidoyl substituents affording preferentially the Z-isomer of 1c. However, heteronucleophiles such as sodium azide, secondary amine and cesium fluoride were unsuccessfully tested. Finally, using the conditions developed by Burkhard and Carreira [26], the opening of the fluoroalkylidene-oxetane ring was investigated with hydrobromic acid (HBr 33 wt % in AcOH, 2.3 equiv) in diethyl ether (Table 1, entry 4). This reaction proved faster and reached completion after 45 min at 20 °C, giving product 1d with an excellent yield of 94% and an E/Z selectivity of 89:11. The isomers could be separated and crystals of the major isomer were obtained by recrystallization. The X-ray diffraction analysis clearly showed that the bromine atom was located on the carbon trans to the fluorine atom resulting in the major product with the E-geometry (see Supporting Information File 1). It should be noted that longer reaction times resulted in slow solvolysis of alcohol 1d with acetic acid giving mainly the corresponding acetate (not shown). Product 1d was also obtained in a similar E/Z ratio (88:12) when the ring-opening reaction was performed in dichloromethane with tetrabutylammonium bromide (TBAB) as the bromide source and boron trifluoride diethyl etherate as an activator (Table 1, entry 5). Nevertheless, the isolated yield of the product decreased to 71%. Next, the reaction performed with HBr/AcOH was extended to alkylidene oxetanes substituted by an alkyl chain, and a pyrimidine base. The presence of the alkyl chain in place of the phthalimido group did not affect the selectivity observed with 1. The ring opening reaction of the n-octyl substituted oxetane 2 resulted in an excellent selectivity of 94:6 towards the E-isomer of bromoalkylated product 2d (Table 1, entry 6). The E/Z mixture of 2d was isolated in moderate yield (53%). In this case the corresponding acetate was observed as a minor product (15%) but with a similar selectivity of 92:8, although it could not be isolated in pure form. Unfortunately, the introduction of a nucleic base such as N3-benzoyliodouracil instead of the phthalimido group gave a complex mixture of products (Table 1, entry 7). In contrast, the ring-opening reaction was successful when performed in the presence of TBAB and BF3·Et2O and afforded the E-alkene product 3d with good selectivity (E/Z ratio > 96:4) and 76% yield (Table 1, entry 8). The geometric assignment of compound 3d was corroborated by 1D NOESY experiments in which after selective irradiation of the protons α to the nitrogen atom, a response was observed only for the protons α to the bromine atom, thus indicating their spatial proximity.
Table 1: Oxetane opening by various nucleophiles.
| entry | oxetane |
Nu
(equiv) |
additive
(equiv) |
t (h) | T (° C) | solvent | product | E/Z ratioa | Yield (%)b |
| 1 | 1 |
MeOH
(20) |
CSA
(1) |
26 | 40 | CH2Cl2 | 1a | 40:60 | 60 |
| 2 | 1 |
BnOH
(24) |
CSA
(1) |
20 | 80 | neat | 1b | 45:55 | n.a. |
| 3 | 1 |
BTSH
(1.4) |
CSA
(1) |
24 | 20 | CH2Cl2 | 1c | 25:75 | 65 |
| 4 | 1 |
HBr
(2.3) |
AcOHc | 0.75 | 20 | Et2O | 1d | 89:11 | 94 |
| 5 | 1 |
TBAB
(2.5) |
BF3·Et2O
(1.1) |
2 | −20 | CH2Cl2 | 1d | 88:12 | 71 |
| 6 | 2 |
HBr
(2.3) |
AcOHc | 0.75 | 20 | CH2Cl2 | 2d | 94:6 | 53 |
| 7 | 3 |
HBr
(2.3) |
AcOHc | 0.75 | 20 | CH2Cl2 | – | – | – |
| 8 | 3 |
TBAB
(2.5) |
BF3·Et2O
(1.1) |
2 | −20 | CH2Cl2 | 3d | 96:4 | 76 |
aDetermined by 19F NMR of the crude mixture; byield of isolated product; cHBr 33 wt % in AcOH solution.
In order to elucidate the selectivity control in this ring-opening reaction displayed by bromide ion despite the presence of bulky substituents (phthalimido and alkyl groups), a comparative study was initiated using non-fluorinated alkylidene oxetanes as substrates. Since the oxetane ring was attacked from the side of the bulky phthalimide or alkyl chain, it appeared plausible that the selectivity observed for the reaction did not originate from steric hindrance. Accordingly, the electronic influence of fluorine was explored. Thus, the non-fluorinated analogues bearing a phthalimido group (4) and an alkyl chain (5) were synthesized, and submitted to the ring-opening reaction [27,28]. Pleasingly, when subjecting compound 4 to the previous reaction conditions, a single isomer of bromoalcohol 4d formed (determined by 1H NMR of the crude) and the product was isolated in an excellent yield of 94% (Table 2, entry 3). The geometry of the product was confirmed to be E-4d by X-ray diffraction analysis (see Supporting Information File 1). The attack of the bromide ion this time occurred from the side of the alkene hydrogen atom (cis attack) and away from the bulky phthalimido group giving solely product E-4d. Likewise, the reaction of the alkyl-substituted substrate 5 proceeded with excellent E-selectivity (as determined by 1H NMR of the crude). As in case of the fluorinated analogue, a substantial part of the newly formed alcohol 5d reacted with acetic acid forming the corresponding acetate 5d’. Interestingly, it appeared that only the E-isomer of 5d reacted with AcOH giving pure E-5d’ in 44% isolated yield. This left an 81:19 E/Z mixture of 5d from which the pure alcohol E-5d could be isolated in 34% yield (Table 2, entry 4). The overall E/Z ratio for the ring-opening reaction of 5 was 91:9. 1D NOESY experiments were performed with alcohol 5d and the outcome indicated that the bromide attack took place away from the bulky alkyl chain, resulting in the observed E-selectivity. These outcomes were opposite to those obtained with the fluorinated substrate series, consistent with steric hindrance governing the non-fluorine containing oxetane reactions but electronics influencing and reversing the regioselectivity of the fluoro-oxetane reactions.
Table 2: Selectivity in the presence or absence of the fluorine atom.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-160-i10.svg?max-width=637&scale=1.0)
|
|||||||
| entry | substrate | R2 | R1 | R3 | product | E/Z ratioa | yield (%)b |
| 1 | 1 | F | phthalimido | H | 1d | 89:11 | 94 |
| 2 | 2 | F | C7H15 | H | 2d | 94:6 | 53 |
| 3 | 4c | H | phthalimido | H | 4d | 100:0 | 94 |
| 4 | 5 | H | C8H17 | H | 5d | 81:19 | 34d |
| Ac | 5d’ | 100:0 | 44 | ||||
aDetermined by 19F NMR (1, 2) or 1H NMR (4, 5) of the crude mixture; byield of isolated product; csubstrate 4 was contaminated with 1 molar equivalent of phthalimide; dyield of isolated E-5d.
The reaction was then extended to fluoroalkylidene-oxetane 8 to expand the range of tetrasubstituted fluoroalkenes accessible via this method (Scheme 2). A variety of conditions were explored to prepare the protected alcohol 8 including an unsuccessful reduction of the corresponding ethyl ester (vide infra, 12). Then we turned our attention to the modified Julia reaction since the reduction of the ester functionality could be achieved at the sulfide stage [29], prior to its oxidation to give 6. Alcohol 6 was not stable in basic medium, as a Smiles rearrangement occurred leading to fluoroethylene and benzothiazolone. Therefore, its benzylation was explored under acidic conditions with benzyl trichloroacetimidate (1.5 equiv) and a catalytic amount of trifluoromethanesulfonic acid. This gave benzyl ether 7 as a 2.5:1 mixture with N-benzylbenzothiazolone (not shown, Scheme 2). After purification, benzyl ether 7 was successfully subjected to the modified Julia olefination conditions with 3-oxetanone, to give the corresponding alkene 8 in 79% yield.
![[1860-5397-16-160-i2]](/bjoc/content/inline/1860-5397-16-160-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of benzyloxy-substituted fluoroethylidene-oxetane derivative 8.
Scheme 2: Synthesis of benzyloxy-substituted fluoroethylidene-oxetane derivative 8.
With alkene 8 in hand, the ring-opening reaction was explored in the presence of hydrobromic acid (HBr 33 wt % in AcOH) in diethyl ether (Table 3). As observed with the phthalimido group, the reaction led to alkene E-9 as the major product, together with alkene Z-9 and a third product that was identified as the 2,5-dihydrofuran derivative 10.
Table 3: Opening of the benzyloxy-substituted fluoroethylideneoxetane derivative 8.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-160-i11.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Nu (equiv) |
Additive
(equiv) |
Solvent |
Reaction
time (h) |
Selectivitya | Yield of 9 (%) | ||
| E-9 | Z-9 | 10 | ||||||
| 1 | HBr (2.3) | AcOH | Et2O | 0.5 | 62 | 8 | 26 | 45 |
| 2 |
HBr (1.2)
TBAB (2.5) |
AcOH | CH2Cl2 | 0.5 | 81 | 15 | 4 | 74b |
| 3 | TBAB (2.5) | BF3·OEt2 (1.1) | CH2Cl2 | 1 | 90 | 10 | 0 | 66 |
aDetermined by 19F NMR of the crude mixture; byield for the 92:8 E/Z mixture.
Using the standardized conditions, but at a temperature of 0 °C instead of 20 °C, for 30 min, complete conversion was achieved and the three products E,Z-9 and 10 were present in a 62:8:26 ratio as determined by 19F NMR, the remaining 4% being attributed to acetylated analogues of 9 (Table 3, entry 1). After purification, two products were obtained as a 95:5 mixture and identified by NMR as the desired bromoalcohols E-9 and Z-9, respectively. The selectivity of the oxetane ring opening (crude E/Z ratio: 89:11) was again governed by the presence of the fluorine atom and not by steric hindrance.
In order to limit the competitive formation of the heterocyclic ether 10, the addition of TBAB as a bromide source was explored. To our delight, after 30 min at 0 °C, the crude 19F NMR showed that only 4% of 10 and 96% of 9 as an 84:16 E/Z mixture had formed (Table 3, entry 2). Separation of the two E/Z isomers proved challenging by column chromatography and a 92:8 mixture of E/Z-9 was obtained in 74% yield. Finally, the reaction performed with TBAB in the presence of BF3·Et2O afforded only alkenes 9 with an excellent E-selectivity and in 66% yield (Table 3, entry 3). In this case, we presume 10 was not obtained because the benzyl ether is not nucleophilic enough to react due to it complexation by BF3·OEt2.
To understand in more detail the competitive formation of linear versus cyclic products in the reaction, methanol was explored as the nucleophile instead of bromide ion (Scheme 3). The reaction (43 h, 20 °C) realized in MeOH as a solvent and in the presence of CSA (1 equiv) afforded a mixture of 10 (85%) and the expected methoxyalcohols 11 (10%, 1:1 E/Z ratio) in addition to starting fluoroalkylidene-oxetane 8 (5%). After purification, compound 10 was isolated in 75% yield.
![[1860-5397-16-160-i3]](/bjoc/content/inline/1860-5397-16-160-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Effect of the medium on the selective formation of derivative 10.
Scheme 3: Effect of the medium on the selective formation of derivative 10.
The results from the acid-catalyzed results support the reaction outcomes which depended on the nucleophile (methanol or bromide ion) (Scheme 4). In the presence of excess bromide ions, the direct intermolecular nucleophilic attack of the oxetane (path a) is preferred leading to bromoalcohol 9 (path a). On the other hand, in the presence of the weaker methanol nucleophile, an intramolecular ring-opening reaction by the benzyl ether oxygen is preferred leading to the 2,5–dihydrofuran 10 (path b).
![[1860-5397-16-160-i4]](/bjoc/content/inline/1860-5397-16-160-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism for the formation of dihydrofuran 10.
Scheme 4: Mechanism for the formation of dihydrofuran 10.
Finally, we turned our attention to a last series of reactions exploring the ring-opening reaction of fluoroalkylidene-oxetane 12 (Table 4).
Table 4: Ring-opening reaction from acetate derivative 12.
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-160-i12.svg?max-width=637&scale=1.0)
|
||||||||||
| entry | Nu (equiv) | additive | solvent | t (h) | T (°C) | selectivitya | product (yield)b | |||
| E-13 | 14 | Z-13 | 15 | |||||||
| 1 | HBr (2.5) | AcOH | Et2O | 3.5 | 20 | – | 87 | 1 | 8 | 14 (58) |
| 2 | HBr (1.2) | AcOH | Et2O | 3.5 | 0 | 3 | 76 | 7 | 10 | – |
| 3 | TBAB (2.5) | CSA (2)c | CH2Cl2 | 30 | 20 | – | 7 | 6 | 79 (8)d | 15 (61)e |
| 4 | TBAB (2) | BF3.OEt2 (1.5) | CH2Cl2 | 4 | –20 | – | – | 78 | 22 |
Z-13 (72)f
15 (21)f |
| 5 | TBAB (1.5) | – | CH2Cl2 | 16 | 20 | – | – | – | – | – |
| 6 | – | BF3·OEt2 (1) | CH2Cl2 | 2 | 20 | – | 100 | – | – | 14 (67) |
aDetermined by 19F NMR of the crude mixture; bisolated yield; cthe 2nd equivalent of CSA was added after 24 h; dchlorinated instead of brominated products; eyield of an 87:8:5 mixture of products 15/19/Z-13; fZ-13 and 15 were obtained as a crude mixture, yields were calculated from the crude mass and 1H NMR.
Given the previous results, in the presence of the ester function we expected the ring-opening reaction to proceed with the formation of additional products to alkenes E-13 and Z-13. In fact, two byproducts formed and were identified as β-hydroxymethyl-α-fluorolactone 14 and β-bromomethyl-α-fluorolactone 15 (Table 4) [30]. This ring expansion has been already reported in the literature from oxetane-containing α,β-unsaturated carbonyl derivatives through a Lewis acid-catalyzed rearrangement [31]. When the previous conditions (HBr/AcOH) were tried (3.5 h, 0 °C to 20 °C), hydroxymethyllactone 14 (87%, Table 4, entry 1) was the main product with traces of the corresponding acetate (4%, not shown), the bromomethyllactone 15 (8%), and alkene Z-13 (1%). Only lactone 14 could be isolated in a pure form (58% yield). The amount of HBr/AcOH had little influence on the selectivity (Table 4, entry 2) of the reaction. However, in the presence of TBAB (2.5 equiv) and CSA (2 equiv) an 84% conversion into mainly brominated lactone 15 (79%) with traces of hydroxylated lactone 14, and of bromoalkene Z-13 was determined by crude 19F NMR. The remaining 8% appeared to be the β-chloromethyllactone 19 (see Scheme 7 below), an analogue of 15 as determined by NMR analysis and supported by HRMS. All three halogenated products were purified and isolated as a mixture (≈61% determined by NMR). The contrasting result observed with HBr/AcOH and TBAB/CSA highlighted the importance of the acidity of the medium on the reaction course of the ring-opening reaction (Scheme 5). The former, using an excess of acid (33% HBr in AcOH solution) favored the direct nucleophilic attack (path b) leading to lactone 14, whereas the latter in the presence of TBAB/CSA allowed bromide addition on the same side of the fluorine atom (path a) leading to lactone 15 [31]. However, the reaction can be stopped at the alcohol stage when performed in the presence of BF3·Et2O instead of CSA to afford alkene Z-13 as the major product. The use of boron trifluoride etherate as an activator combined with TBAB afforded exclusively alkene Z-13 as evidenced by TLC, but after work-up, 22% of the bromomethyllactone 15 was observed (Table 4, entry 4). It appeared obvious that Z-13 could cyclize under acidic conditions and during the purification gave β-bromomethyllactone 15. This was later confirmed when various ester/lactone mixtures obtained after the ring-opening reaction were treated with acid (PTSA) in Et2O giving pure lactone 15 or at least enriched mixtures depending on the reaction times (Scheme 5).
![[1860-5397-16-160-i5]](/bjoc/content/inline/1860-5397-16-160-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanism for the formation of unsaturated lactones 14 and 15.
Scheme 5: Mechanism for the formation of unsaturated lactones 14 and 15.
As a control, 12 was shown to be unreactive to TBAB on its own (Table 4, entry 5). Once again, the effect of the fluorine atom was highlighted by an investigation with the non-fluorinated alkylidene oxetane 16. The latter was subjected to the HBr/AcOH ring-opening reaction conditions (Scheme 6). Remarkably, the cyclic products β-hydroxymethyl and β-bromomethyl-γ-lactones 17 and 18 were obtained in an 8:92 ratio [32]. This complete reversal of selectivity in comparison with fluoroalkylidene-oxetane 12, where the β-hydroxymethyl-γ-lactone 14 was obtained, confirmed an electronic influence of the fluorine atom on these ring-opening reactions (Table 3, entries 1 and 2). In the case of fluoroalkylidene-oxetane 12 and, in contrast with 16, when subjected to HBr/AcOH, the electronic repulsion induced between fluorine and bromine limited the intermolecular ring-opening reaction by bromide in favor of a faster intramolecular reaction involving the ester group leading to 14. Indeed, a competitive cyclization reaction occurred forming 14 with HBr/AcOH and confirmed when the reaction was performed in the presence of BF3·Et2O only (Table 4, entries 1, 2, and 6). Of note in contrast to 8, when TBAB/BF3·Et2O was added we cannot exclude a control by steric or electronic repulsions between bromide ions and the ester function leading to Z-13 and 15 instead of expected alkene E-13 and to the lactone 18 from alkylidene oxetanes 12 and 16, respectively.
![[1860-5397-16-160-i6]](/bjoc/content/inline/1860-5397-16-160-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Opening reaction of ethyl 2-(oxetanyl-3-idene)acetate (16).
Scheme 6: Opening reaction of ethyl 2-(oxetanyl-3-idene)acetate (16).
Having established selective approaches for the preparation of halogenated allylic fluoroalkenes, their use in the synthesis of highly functionalized tetrasubstituted fluoroalkenes was explored. The derivatization of the three brominated products, lactone 15 and alkenes E-1d and E-9, was studied either on the bromomethyl (CH2Br) or on the hydroxymethyl (CH2OH) arm, when applicable.
First, from a mixture of lactones 15 and 19 substitution on the bromomethyl arm was performed using sodium azide (Scheme 7). The reaction proceeded smoothly in DMF but it proved difficult to extract product 20 from water. When the reaction was performed in acetone this allowed for a simple filtration of the sodium chloride and bromide salts formed and resulted in very satisfactory yields (91–97%) after column chromatography. An Arbuzov phosphonylation was performed on the crude lactone product 15 (containing 10% of ester Z-13) and proved successful with phosphonolactone 21 being isolated in 77% yield after column chromatography (Scheme 7). To access tetrasubstituted alkenes, reduction of 21 to generate diol 22 was explored with lithium borohydride in Et2O. However, the reaction was slow at 20 °C and did not progress beyond 50% conversion even after the addition of excess LiBH4. After purification by flash chromatography starting lactone 21 was obtained in 49% yield and the desired diol 22 in 47% yield. When the reaction was carried out in refluxing THF, a complete conversion was achieved but also with impurities. This route was not investigated further, and instead functionalization of alkene E-1d was explored.
![[1860-5397-16-160-i7]](/bjoc/content/inline/1860-5397-16-160-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Functionalization of bromomethyllactone 15 and its analogues.
Scheme 7: Functionalization of bromomethyllactone 15 and its analogues.
Direct ring-opening reactions of fluoroalkylidene-oxetane 1 with heteronucleophiles were previously explored and success was only possible with thiols, such as mercaptobenzothiazole (Table 1). However, the reaction was not selective and afforded a mixture of E/Z alkenes 1c. The functionalization of alkene E-1d via displacement of the bromine atom (Scheme 8), with nucleophiles such as CsF and NaN3 was then studied. When the reaction was performed in DMF products E-23 and Z-24 were generated in 92% and 93% yield, respectively. Reactions with amines and thiols such as pyrrolidine and 2-mercaptobenzothiazole, gave rise to the products Z-25 and E-1c in 96% yield, respectively. These reactions were carried out in dichloromethane in the presence of Et3N. It should be noted that following this two-step method, pure E-1c could be obtained while direct ring opening of 1 with BTSH and CSA resulted in a 25:75 mixture of E/Z-1c. A crystallographic analysis of crystals of Z-25 confirmed the nature and geometry of the obtained product (see Supporting Information File 2). Addition of carbanions or alcoholates was also attempted but the starting bromide E-1d degraded under these conditions.
![[1860-5397-16-160-i8]](/bjoc/content/inline/1860-5397-16-160-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Functionalization by substitution reaction of the bromide E-1d vs ring-opening reaction of the oxetane 1.
Scheme 8: Functionalization by substitution reaction of the bromide E-1d vs ring-opening reaction of the oxet...
Finally, this expeditious synthesis of tetrasubstituted fluoroalkenes by sequential ring-opening and nucleophilic substitution reactions was applied to test the robustness of a selective preparation of precursors of ACN (VII) bearing different functional groups (Scheme 9). A particular focus was applied to the preparation of the phosphonate 29, a precursor of VII that is not accessible from diol VIII.
![[1860-5397-16-160-i9]](/bjoc/content/inline/1860-5397-16-160-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Preparation of tetrasubstituted fluoroalkenes.
Scheme 9: Preparation of tetrasubstituted fluoroalkenes.
First, starting from pure alkene E-9, the introduction of a protected alcohol as a mimic of the naturally occurring 3’-hydroxy group was achieved by allylic bromine displacement with AcOK to efficiently afford alkene 26. The phosphonate was introduced in three steps through the formation of intermediate mesylate 27. This mesylate was progressed without purification, albeit contaminated (10%) with the corresponding chloride (not shown). An Arbuzov reaction was performed directly on the allylic bromide obtained by treatment of 27 with LiBr (5 equiv), to give the phosphonate 29 in 76% overall yield. Finally, azide 28 was obtained in 89% yield in two steps from the non-isolated intermediate mesylate 27. After deacetylation, 28 was readily converted to E-24 (see Scheme 8). These transformations of alkene E-9 illustrated how the geometry can be controlled for the preparation of tetrasubstituted fluoroalkenes. The synthesis of nucleotide mimics from either phosphonate 29 or azide 28 is underway and will be reported in due course.
Conclusion
The selective synthesis of tetrasubstituted E- or Z-fluoroalkenes was achieved by ring-opening reactions of fluoroalkylidene-oxetanes, with the presence of the fluorine atom governing regioisomeric attack of the bromide ion. Functionalization of the resultant bromoalcohols with nucleophiles led, in two steps from oxetanes, to a series of highly functionalized tetrasubstituted fluoroalkenes with excellent geometric control. This method offers ready access to novel fluoroalkenes as potential precursors of important drug mimics.
Supporting Information
The experimental section describing the preparation of all new compounds, the copies of the NMR data (1H NMR, 13C NMR, 19F NMR), HOESY and NOESY experiments and crystallographic data for compounds 1d, 4d and 25. The CIF files of 1d, 4d and 25.
| Supporting Information File 1: Experimental section and copies of spectra. | ||
| Format: PDF | Size: 8.0 MB | Download |
| Supporting Information File 2: Crystallographic data (cif) for compounds E-1d, E-4d, and Z-25. | ||
| Format: ZIP | Size: 415.5 KB | Download |
References
-
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003
Return to citation in text: [1] -
Len, C.; Mackenzie, G. Tetrahedron 2006, 62, 9085–9107. doi:10.1016/j.tet.2006.07.050
Return to citation in text: [1] -
Bassetto, M.; Slusarczyk, M. Pharm. Pat. Anal. 2018, 7, 277–299. doi:10.4155/ppa-2018-0028
Return to citation in text: [1] -
Xie, M.-S.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Tetrahedron Lett. 2014, 55, 7156–7166. doi:10.1016/j.tetlet.2014.11.060
Return to citation in text: [1] -
De Clercq, E. Med. Res. Rev. 2013, 33, 1278–1303. doi:10.1002/med.21283
Return to citation in text: [1] -
Schramm, V. L. Chem. Rev. 2018, 118, 11194–11258. doi:10.1021/acs.chemrev.8b00369
Return to citation in text: [1] -
Kasthuri, M.; El Amri, C.; Lefort, V.; Périgaud, C.; Peyrottes, S. New J. Chem. 2014, 38, 4736–4742. doi:10.1039/c4nj00813h
Return to citation in text: [1] -
Topalis, D.; Pradère, U.; Roy, V.; Caillat, C.; Azzouzi, A.; Broggi, J.; Snoeck, R.; Andrei, G.; Lin, J.; Eriksson, S.; Alexandre, J. A. C.; El-Amri, C.; Deville-Bonne, D.; Meyer, P.; Balzarini, J.; Agrofoglio, L. A. J. Med. Chem. 2011, 54, 222–232. doi:10.1021/jm1011462
Return to citation in text: [1] [2] -
Amblard, F.; Nolan, S. P.; Schinazi, R. F.; Agrofoglio, L. A. Tetrahedron 2005, 61, 537–544. doi:10.1016/j.tet.2004.11.019
Return to citation in text: [1] -
Varada, M.; Erande, N. D.; Kumar, V. A. RSC Adv. 2015, 5, 97824–97830. doi:10.1039/c5ra15673d
Return to citation in text: [1] -
Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306–322. doi:10.1055/s-0034-1379537
Return to citation in text: [1] -
Choi, M.-H.; Lee, C.-K.; Jeong, L. S.; Chun, M. W.; Kim, H.-D. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 681–684. doi:10.1081/ncn-100002350
Return to citation in text: [1] -
Choi, M.-H.; Kim, H.-D. Arch. Pharmacal Res. 1997, 20, 501–506. doi:10.1007/bf02973948
Return to citation in text: [1] [2] -
Prunier, A.; Calata, C.; Legros, J.; Maddaluno, J.; Pfund, E.; Lequeux, T. J. Org. Chem. 2013, 78, 8083–8097. doi:10.1021/jo401356j
Return to citation in text: [1] -
Drouin, M.; Hamel, J.-D.; Paquin, J.-F. Synthesis 2018, 50, 881–955. doi:10.1055/s-0036-1591867
Return to citation in text: [1] -
Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Chem. Soc. Rev. 2011, 40, 2867–2908. doi:10.1039/c0cs00201a
Return to citation in text: [1] -
Yanai, H.; Taguchi, T. Eur. J. Org. Chem. 2011, 5939–5954. doi:10.1002/ejoc.201100495
Return to citation in text: [1] -
Pfund, E.; Lequeux, T.; Gueyrard, D. Synthesis 2015, 47, 1534–1546. doi:10.1055/s-0034-1380548
Return to citation in text: [1] -
Laporte, R.; Prunier, A.; Pfund, E.; Roy, V.; Agrofoglio, L. A.; Lequeux, T. Eur. J. Org. Chem. 2015, 3121–3128. doi:10.1002/ejoc.201500172
Return to citation in text: [1] -
Yadav, J. S.; Singh, V. K.; Srihari, P. Org. Lett. 2014, 16, 836–839. doi:10.1021/ol403604u
Return to citation in text: [1] -
Burkhard, J. A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 9052–9067. doi:10.1002/anie.200907155
Return to citation in text: [1] -
Wang, Z.; Chen, Z.; Sun, J. Angew. Chem., Int. Ed. 2013, 52, 6685–6688. doi:10.1002/anie.201300188
Return to citation in text: [1] -
Burkhard, J.; Carreira, E. M. Org. Lett. 2008, 10, 3525–3526. doi:10.1021/ol801293f
Return to citation in text: [1] -
Jaśkowska, J.; Kowalski, P. J. Heterocycl. Chem. 2008, 45, 1371–1375. doi:10.1002/jhet.5570450519
Return to citation in text: [1] -
Wolin, R.; Connolly, M.; Afonso, A.; Hey, J. A.; She, H.; Rivelli, M. A.; Willams, S. M.; West, R. E., Jr. Bioorg. Med. Chem. Lett. 1998, 8, 2157–2162. doi:10.1016/s0960-894x(98)00379-5
Return to citation in text: [1] -
Calata, C.; Pfund, E.; Lequeux, T. J. Org. Chem. 2009, 74, 9399–9405. doi:10.1021/jo901540c
Return to citation in text: [1] -
Fort, D. A.; Woltering, T. J.; Alker, A. M.; Bach, T. Heterocycles 2014, 88, 1079–1100. doi:10.3987/com-13-s(s)67
Return to citation in text: [1] -
White, A. R.; Kozlowski, R. A.; Tsai, S.-C.; Vanderwal, C. D. Angew. Chem., Int. Ed. 2017, 56, 10525–10529. doi:10.1002/anie.201704119
Return to citation in text: [1] [2] -
Fort, D. A.; Woltering, T. J.; Nettekoven, M.; Knust, H.; Bach, T. Chem. Commun. 2013, 49, 2989–2991. doi:10.1039/c3cc40757h
Return to citation in text: [1]
| 31. | White, A. R.; Kozlowski, R. A.; Tsai, S.-C.; Vanderwal, C. D. Angew. Chem., Int. Ed. 2017, 56, 10525–10529. doi:10.1002/anie.201704119 |
| 30. | Fort, D. A.; Woltering, T. J.; Alker, A. M.; Bach, T. Heterocycles 2014, 88, 1079–1100. doi:10.3987/com-13-s(s)67 |
| 31. | White, A. R.; Kozlowski, R. A.; Tsai, S.-C.; Vanderwal, C. D. Angew. Chem., Int. Ed. 2017, 56, 10525–10529. doi:10.1002/anie.201704119 |
| 1. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 2. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 3. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 4. | O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003 |
| 11. | Topalis, D.; Pradère, U.; Roy, V.; Caillat, C.; Azzouzi, A.; Broggi, J.; Snoeck, R.; Andrei, G.; Lin, J.; Eriksson, S.; Alexandre, J. A. C.; El-Amri, C.; Deville-Bonne, D.; Meyer, P.; Balzarini, J.; Agrofoglio, L. A. J. Med. Chem. 2011, 54, 222–232. doi:10.1021/jm1011462 |
| 12. | Amblard, F.; Nolan, S. P.; Schinazi, R. F.; Agrofoglio, L. A. Tetrahedron 2005, 61, 537–544. doi:10.1016/j.tet.2004.11.019 |
| 13. | Varada, M.; Erande, N. D.; Kumar, V. A. RSC Adv. 2015, 5, 97824–97830. doi:10.1039/c5ra15673d |
| 27. | Jaśkowska, J.; Kowalski, P. J. Heterocycl. Chem. 2008, 45, 1371–1375. doi:10.1002/jhet.5570450519 |
| 28. | Wolin, R.; Connolly, M.; Afonso, A.; Hey, J. A.; She, H.; Rivelli, M. A.; Willams, S. M.; West, R. E., Jr. Bioorg. Med. Chem. Lett. 1998, 8, 2157–2162. doi:10.1016/s0960-894x(98)00379-5 |
| 10. | Kasthuri, M.; El Amri, C.; Lefort, V.; Périgaud, C.; Peyrottes, S. New J. Chem. 2014, 38, 4736–4742. doi:10.1039/c4nj00813h |
| 29. | Calata, C.; Pfund, E.; Lequeux, T. J. Org. Chem. 2009, 74, 9399–9405. doi:10.1021/jo901540c |
| 7. | Xie, M.-S.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Tetrahedron Lett. 2014, 55, 7156–7166. doi:10.1016/j.tetlet.2014.11.060 |
| 8. | De Clercq, E. Med. Res. Rev. 2013, 33, 1278–1303. doi:10.1002/med.21283 |
| 9. | Schramm, V. L. Chem. Rev. 2018, 118, 11194–11258. doi:10.1021/acs.chemrev.8b00369 |
| 24. | Burkhard, J. A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 9052–9067. doi:10.1002/anie.200907155 |
| 25. | Wang, Z.; Chen, Z.; Sun, J. Angew. Chem., Int. Ed. 2013, 52, 6685–6688. doi:10.1002/anie.201300188 |
| 5. | Len, C.; Mackenzie, G. Tetrahedron 2006, 62, 9085–9107. doi:10.1016/j.tet.2006.07.050 |
| 6. | Bassetto, M.; Slusarczyk, M. Pharm. Pat. Anal. 2018, 7, 277–299. doi:10.4155/ppa-2018-0028 |
| 26. | Burkhard, J.; Carreira, E. M. Org. Lett. 2008, 10, 3525–3526. doi:10.1021/ol801293f |
| 16. | Choi, M.-H.; Kim, H.-D. Arch. Pharmacal Res. 1997, 20, 501–506. doi:10.1007/bf02973948 |
| 22. | Laporte, R.; Prunier, A.; Pfund, E.; Roy, V.; Agrofoglio, L. A.; Lequeux, T. Eur. J. Org. Chem. 2015, 3121–3128. doi:10.1002/ejoc.201500172 |
| 15. | Choi, M.-H.; Lee, C.-K.; Jeong, L. S.; Chun, M. W.; Kim, H.-D. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 681–684. doi:10.1081/ncn-100002350 |
| 16. | Choi, M.-H.; Kim, H.-D. Arch. Pharmacal Res. 1997, 20, 501–506. doi:10.1007/bf02973948 |
| 17. | Prunier, A.; Calata, C.; Legros, J.; Maddaluno, J.; Pfund, E.; Lequeux, T. J. Org. Chem. 2013, 78, 8083–8097. doi:10.1021/jo401356j |
| 23. | Yadav, J. S.; Singh, V. K.; Srihari, P. Org. Lett. 2014, 16, 836–839. doi:10.1021/ol403604u |
| 14. | Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306–322. doi:10.1055/s-0034-1379537 |
| 32. | Fort, D. A.; Woltering, T. J.; Nettekoven, M.; Knust, H.; Bach, T. Chem. Commun. 2013, 49, 2989–2991. doi:10.1039/c3cc40757h |
| 11. | Topalis, D.; Pradère, U.; Roy, V.; Caillat, C.; Azzouzi, A.; Broggi, J.; Snoeck, R.; Andrei, G.; Lin, J.; Eriksson, S.; Alexandre, J. A. C.; El-Amri, C.; Deville-Bonne, D.; Meyer, P.; Balzarini, J.; Agrofoglio, L. A. J. Med. Chem. 2011, 54, 222–232. doi:10.1021/jm1011462 |
| 18. | Drouin, M.; Hamel, J.-D.; Paquin, J.-F. Synthesis 2018, 50, 881–955. doi:10.1055/s-0036-1591867 |
| 19. | Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Chem. Soc. Rev. 2011, 40, 2867–2908. doi:10.1039/c0cs00201a |
| 20. | Yanai, H.; Taguchi, T. Eur. J. Org. Chem. 2011, 5939–5954. doi:10.1002/ejoc.201100495 |
| 21. | Pfund, E.; Lequeux, T.; Gueyrard, D. Synthesis 2015, 47, 1534–1546. doi:10.1055/s-0034-1380548 |
© 2020 Fontenelle et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)