Abstract
The hydrofluorocarbon 245 isomers, 1,1,1,3,3-pentafluoropropane, 1,1,1,2,2- pentafluoropropane, and 1,1,1,2,3-pentafluoropropane (HFC-245fa, HFC-245cb, and HFC-245eb) were activated through C–F bond activations using aluminium chlorofluoride (ACF) as a catalyst. The addition of the hydrogen source Et3SiH is necessary for the activation of the secondary and tertiary C–F bonds. Multiple C–F bond activations such as hydrodefluorinations and dehydrofluorinations were observed, followed by hydroarylation and Friedel–Crafts-type reactions under mild conditions.

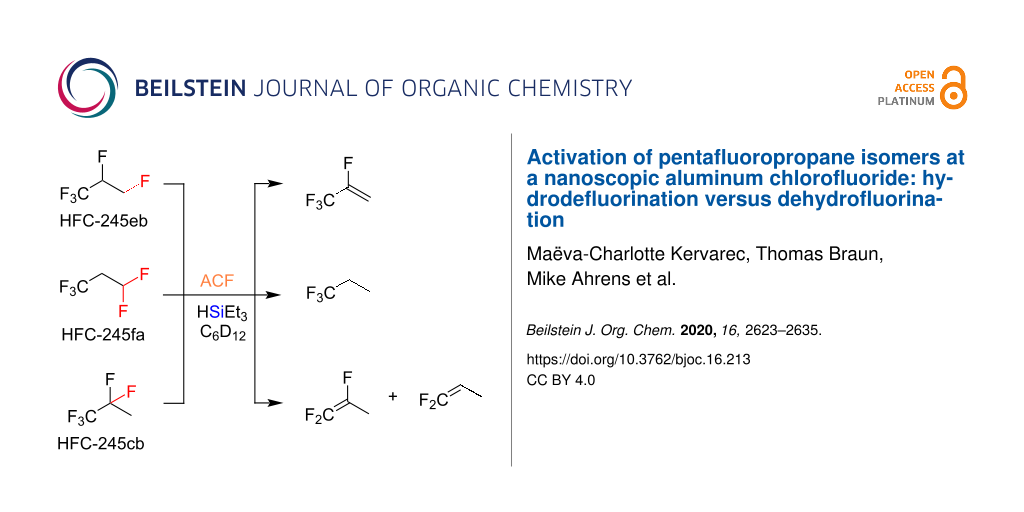
Graphical Abstract
Introduction
Hydrofluorocarbons (HFCs) have been intensively used in daily life, mainly due to their excellent properties in refrigeration applications [1-3]. In the past, HFCs were considered as replacements that do not deplete ozone for chlorofluorocarbons (CFCs) and hydrochlorofluorocarbons (HCFCs), which have been strictly regulated by the Montreal protocol [4-6]. However, due to the high global warming potential (GWP), HFCs have also been included in the Montreal protocol in 2019 (Kigali amendment) and have to be phased out [7-10].
On the other hand, HFCs are valuable starting compounds or intermediate products for the synthesis of hydrofluoroolefins (HFOs), which have been regarded as the next generation of refrigerants, exhibiting zero ozone depletion potential (ODP) and a negligible GWP [11-13]. A considerable amount of studies has been carried out to synthesize HFOs under mild conditions [11,14-16]. Among them are routes to access 2,3,3,3-tetrafluoropropene and 1,3,3,3-tetrafluoropropene (HFO-1234yf and HFO-1234ze), for which numerous patents suggest synthetic pathways and showcase the reactivity [12,13,15]. One possibility for the preparation includes the conversion of pentafluoropropanes (HFC-245 isomers) using chromia-based catalysts, or metal chloride/fluoride (AlF3, MgF2)-supported catalysts at elevated temperatures (350 °C) [11,14,15,17,18]. The group of Lu recently reported the gas-phase transformation of 1,1,1,3,3-pentafluoropropane (HFC-245eb) into 1,3,3,3-tetrafluoropropene (HFO-1234ze) using mesoporous nanoscopic aluminum fluoride-based catalysts [19]. The catalysts were prepared via a sol–gel process in the presence of polyols, allowing for the evolution of a large surface area and improved acidic properties when compared to fluorinated Cr2O3 or traditional β-AlF3 catalysts. At a reaction temperature set at 280 °C, the conversion of 1,1,1,2,2- pentafluoropropane (HFC-245fa) into the 1,3,3,3-tetrafluoropropene (HFO-1234ze) varied between 50 and 60%, depending on the conditions used to synthesize the catalyst, reaching almost full selectivity. The harsh conditions are in part needed due to the high dissociation energy of C–F bonds, and in general, C–F activation steps are considered to be challenging [20-27].
Solid Lewis acids with a high fluoride ion affinity as catalysts are useful tools for C–F bond activation reactions since the Lewis acidic centers can induce dehydrofluorination reactions, involving the abstraction of a fluoride ion by heterolytic bond cleavage [28-31]. AlF3-based catalysts are among the strongest Lewis acidic materials. They exhibit an effective activity in C–F bond conversion reactions and are widely investigated [16,28,32-39]. Especially microporous aluminum chlorofluoride (ACF, AlClxF3−x; x = 0.05–0.3), which has a large surface area (>200 m2g−1) and was patented by Dupont in 1992, has been extensively studied [40-46]. It is an amorphous aluminum fluoride doped with chlorine atoms which causes a distortion of the structure resulting in the amorphicity and high Lewis acidity of the compound. The reactivity of ACF towards C–F bond activations was deeply investigated. For instance, the activation of fluoromethanes was observed at ACF in the presence of HSiEt3 as a hydrogen source to produce, in the presence of benzene as the solvent, Friedel–Crafts products as main compounds [47]. In contrast, the hydrodefluorination products were generated in the absence of benzene. Thermodynamically, the generation of strong H–F, Al–F, or Si–F bonds can enforce an activation of C–F bonds under mild conditions, and hence the addition of the silane HSiEt3 as a hydrogen source [27,48,49]. More recently, ACF was shown to efficiently convert the fluoroalkenes HFO-1234yf (1) and HFO-1234ze (4a) in the presence of the hydrogen source HSiEt3 into the hydrodefluorination or Friedel–Crafts products (Scheme 1) [16].
![[1860-5397-16-213-i1]](/bjoc/content/inline/1860-5397-16-213-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactivity of tetrafluoropropanes HFO-1234yf (1) (top) and HFO-1234ze (4a) (bottom) in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source [16].
Scheme 1: Reactivity of tetrafluoropropanes HFO-1234yf (1) (top) and HFO-1234ze (4a) (bottom) in the presence...
The activation of fluoropentane was achieved using a modified ACF, loaded with germane or silane [39]. When silane was immobilized at the surface of ACF in the presence of benzene, Friedel–Crafts products were again generated. In comparison, when ACF was loaded with germane, dehydrofluorination products were detected. Besides, 2-chloro-1,1,1,2-tetrafluoropropane (HCFC-244bb) was as well effectively activated at ACF to yield the corresponding dehydrofluorination product 2-chloro-3,3,3-trifluoropropene (HFO-1233xf) without the use of any additional hydrogen source [50]. In the presence of silane and ACF, HFO-1233xf was further activated via allylic hydrodefluorination.
In this paper, we report on the reactivity of ACF towards hydrofluorocarbons, and in particular, the pentafluoropropane isomers (HFC-245). Effective hydrodefluorination and dehydrofluorination steps of pentafluoropropane isomers in the presence of Et3SiH as a hydrogen source at mild conditions are described.
Results and Discussion
Activation of 1,1,1,2,3-pentafluoropropane (HFC-245eb, 10a)
The treatment of 1,1,1,2,3-pentafluoropropane (10a) with ACF at 70 °C in C6D12 gave the dehydrofluorination product 2,3,3,3-tetrafluoropropene (HFO-1234yf, 1) and the isomerization product 1,1,1,2,2-pentafluoropropane (HFC-245cb, 10b) in a 1:2 ratio (Scheme 2, top) with almost full conversion. The group of Kemnitz previously showed that 1 and 10b can be in an equilibrium when HF is present in the reaction mixture [33]. It was demonstrated that starting from 2-chloro-3,3,3-trifluoropropene (HFO-1233xf) in the presence of fluorinated Cr2O3 as a catalyst and HF, 2,3,3,3-tetrafluoropropene (HFO-1234yf, 1) is generated by the replacement of the chlorine substituent with a fluorine atom, and is further transformed by HF addition into 1,1,1,2,2-pentafluoropropane (HFC-245cb, 10b) [33].
![[1860-5397-16-213-i2]](/bjoc/content/inline/1860-5397-16-213-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reactivity of 10a in the presence of ACF as the catalyst in C6D12 (top) or C6D6 (bottom) as solvents.
Scheme 2: Reactivity of 10a in the presence of ACF as the catalyst in C6D12 (top) or C6D6 (bottom) as solvent...
When the aromatic solvent C6D6 was used instead of C6D12, 10a was once more transformed into 1 as the main compound, together with traces of the Friedel–Crafts product CF3CFHCH2C6D5 (11) and the hydroarylation product CF3CFDCH2C6D5 (12) (Scheme 2, bottom), with only 22% conversion. The low conversion in C6D6 could be a consequence of a possible interaction of the aromatic solvent with the surface of ACF, which would result in the blocking of the acidic sites, and thus hamper the adsorption of the substrates. Indeed, in a previous study, a pulse TA experiment suggested the presence of a strong interaction between benzene and the surface of ACF [38]. This result was further confirmed by 1H MAS NMR spectroscopy.
Note, that 10a was activated under mild conditions without the use of an additional hydrogen source, which often has been added for the activation of C–F bonds at ACF [16,39,47]. Several patents cover the transformation of 10a by dehydrofluorination at chromia-based catalysts, but the reaction temperatures were above 200 °C [17,51,52].
Mechanistically, an abstraction of a fluorine from the CH2F group by the surface of ACF can occur, generating carbenium-like species and surface fluorides (Scheme 3). Via HF elimination, the olefin 1 can be produced, followed by a refluorination of the double bond by the released HF, generating 10b (Scheme 3, left). In the presence of C6D6, the hydroarylation product 12 can be generated from 1 at the ACF surface. Alternatively, the aromatic solvent can also attack the carbenium-like species, producing a zwitterionic Wheeland intermediate, which can release the Friedel–Crafts product 11 and DF to regenerate the catalyst (Scheme 3, right).
![[1860-5397-16-213-i3]](/bjoc/content/inline/1860-5397-16-213-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed catalytic cycle of the transformation of 10a in C6D12 and C6D6 in the presence of ACF as the catalyst.
Scheme 3: Proposed catalytic cycle of the transformation of 10a in C6D12 and C6D6 in the presence of ACF as t...
Although a hydrogen source was not needed to accomplish the activation of 10a, it was of interest to introduce a silane, because as mentioned above, recent reports showed that the activation of various substrates treated with ACF was indeed promoted by the presence of silanes [16,39,47]. Thus, the experiments were also conducted in the presence of HSiEt3, either in a solvent (C6D6 or C6D12), or in neat silane under similar conditions (all reactions were carried out at 70 °C and monitored for 7 days).
The treatment of 10a and HSiEt3 in C6D12 generated 1 and 10b again, in addition to traces of 1,1,2-trifluoropropene (2, Scheme 4, top). The ratio between the olefin 1 and the refluorination product 10b observed in the presence of HSiEt3 was 3:1, whereas, without the silane, a ratio of 1:2 was detected. This difference in the ratio might relate to the amount of HF present in the reaction mixture, which would be lower in the presence of silane because the latter can convert with HF into fluorosilane and H2 [39,50,53]. Consequently, less refluorination takes place, and a higher selectivity towards the formation of the olefin 1 is observed. In C6D6, the activation of 10a gave comparable results as when no HSiEt3 was introduced (Scheme 4, middle). Compound 10a is transformed into 1, 11, and 12 with the additional presence of traces of the Friedel–Crafts product CF2=CFCH2C6D5 (3). However, in neat silane, 10a was converted into 1, 10b, 2, 1,1,1,2-tetrafluoropropane (13), 1,1-difluoropropene (5), and 1,1,1-trifluoropropane (6, Scheme 4, bottom). Thus, by having a large excess of silane, a consecutive reactivity was observed, which led to the formation of the hydrodefluorination product 6 as the main compound. However, the reaction is unselective, and various intermediates are still present in considerable amounts (Scheme 4).
![[1860-5397-16-213-i4]](/bjoc/content/inline/1860-5397-16-213-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactivity of 10a in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (top) or C6D6 (middle) as solvents or in neat silane (bottom).
Scheme 4: Reactivity of 10a in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (...
Note, that for the transformation in the presence of silane, the conversions reached over 99% after 7 days at 70 °C, which underlines the significant role of the silane in the reaction mixture. The improved conversion can generally arise from an interaction of silane with the surface of ACF, also competing with the above-mentioned benzene interaction. Furthermore, in the presence of silane, additional mechanistic C–F activation steps have to be considered (Scheme 5). Basically, the immobilization of silane at the Lewis-acidic surface might result in silylium-like species, which subsequently initiate the C–F bond activation at the primary carbon–fluorine bond in 10a, generating FSiEt3, the corresponding carbenium-like species, and a surface-bound hydride. At this stage, either the hydrodefluorination product 13 can be generated, or dehydrofluorination occurs to furnish the olefin 1 and H2, both in the presence of silane. Additionally, 1 can further react with any silylium ion species at the surface of ACF, resulting in a C–F bond cleavage at the CF3 group, yielding once again a surface hydride and the corresponding carbenium ion. Subsequently, the allylic hydrodefluorination product 2 is formed. Allylic hydrodefluorination reactions were previously observed at ACF. Indeed, in the presence of silane and ACF, the CF3 group in tetrafluoropropenes (HFO-1234yf, 1 and HFO-1234ze, 4) was transformed into an olefinic CF2 group (Scheme 1) [16]. Previous MAS NMR studies also gave evidence for the existence of silylium species at an ACF surface [39,47]. In addition, silylium species that are stabilized by weakly coordinating anions can also catalyze hydrodefluorination reactions in a homogeneous phase with silanes as hydrogen source [54-57]. In contrast, silylium-mediated dehydrofluorination reactions have not been found in a homogeneous phase, but germylium ions can promote such reaction pathways [58]. Nevertheless, the formation of the compounds 1, 10b, 11, and 12 can alternatively be initiated by the Lewis acidity of ACF itself, as outlined above without the presence of silane (see Scheme 3). Therefore, as an alternative to the initial formation of the surface silylium ion species at ACF, it is in principle also conceivable that carbenium species can be initially produced by an abstraction of a fluoride ion from a fluorinated group by the surface of the catalyst. Then, carbenium ions can react with silane to yield hydrodefluorination products, or after dehydrofluorination, HF that in the presence of silane, produces FSiEt3 and H2 [39,50]. Furthermore, any intermediate carbenium species, generated directly at the ACF surface or via interaction with a silylium species, can be engaged in Friedel–Crafts-like reactions to give with C6D6 11 or 3, which is consistent with previous studies [16].
![[1860-5397-16-213-i5]](/bjoc/content/inline/1860-5397-16-213-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed catalytic cycle for sylilium-mediated hydrodefluorinations and dehydrofluorinations from 10a at the surface of ACF.
Scheme 5: Proposed catalytic cycle for sylilium-mediated hydrodefluorinations and dehydrofluorinations from 1...
The formation of the products described above (Scheme 4) involves consecutive reaction steps, such as dehydrofluorination, hydrodefluorination, hydrofluorination, allylic defluorination, hydroarylation, and Friedel–Crafts reactions. In the presence of an excess of silane, the dehydrofluorination product 1 and the hydrodefluorination intermediate 13 are generated simultaneously to further lead to 2, 5, and 6 as the main compounds. To get further insight, independent reactions were performed to elucidate reaction patterns and to demonstrate the conceivable transformations between certain products, which were observed in the activation of 10a in the presence of ACF and HSiEt3.
It turned out that the tetrafluoropropene 13 reacts in the presence of silane with ACF as the catalyst in C6D12 or neat silane to give 5 and 6 (ratio 1:2, Scheme 6, top). When C6D6 was used as a solvent in the presence of silane, 5, 14, 9, and the Friedel–Crafts product CF3CH2CH2C6D5 were generated. Note, that in this context, in the absence of silane and in C6D12, the selective formation of 14 was detected (Scheme 6, bottom). In benzene, 14 was also formed as the main compound, together with the corresponding Friedel–Crafts and hydroarylation products, both observed in traces. This suggests that silane promotes the generation of 5 and 6, but for the formation of 14 it is not essential.
![[1860-5397-16-213-i6]](/bjoc/content/inline/1860-5397-16-213-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactivity of 13 in the presence of ACF as the catalyst, with (top) or without (bottom) HSiEt3 as a hydrogen source.
Scheme 6: Reactivity of 13 in the presence of ACF as the catalyst, with (top) or without (bottom) HSiEt3 as a...
Furthermore, another independent reaction by treating 5 with HF was performed (Scheme 7, top), but the hydrofluorination product 6 was only detected in small amounts. In accordance with this result, the treatment of 6 in the presence of silane and ACF gave the dehydrofluorination product 5. The observations nevertheless suggest the presence of an equilibrium between 5 and 6, that depends on the amount of HF or HSiEt3 present in the reaction mixture.
![[1860-5397-16-213-i7]](/bjoc/content/inline/1860-5397-16-213-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Independent reactions starting from 5, 6, or 14 in the presence of ACF as the catalyst.
Scheme 7: Independent reactions starting from 5, 6, or 14 in the presence of ACF as the catalyst.
Additionally, it is conceivable that 14 can further react to form 5 via allylic hydrodefluorination [16]. Therefore, the reactivity of 14 was tested in the presence of ACF and silane in C6D12 or C6D6 (Scheme 7, bottom). Indeed, in C6D12, allylic defluorination to yield 5 was observed, together with 6 and traces of 2-chloro-1,1,1-trifluoropropene (15). The chlorinated product 15 could stem from a plausible HCl formation if the substrate is fluorinating ACF [41]. It should be noted that the synthesis of ACF itself consists of the fluorination of AlCl3 by chlorofluoroalkanes [40,41]. Nevertheless, in C6D6, the formation of 5 and 6 and the Friedel–Crafts product CF2=CHCH2C6D5 (9) was observed from 14.
Based on these findings, a general scheme can be drafted to illustrate the sequential generation of products starting from 10a for the conversions in the presence of silane at ACF (Scheme 8). Note, that the reaction of 1 in the presence of silane, ACF, and solvents was repeated under the same conditions as for the activation of 10a, and the generation of 2 and 3 was confirmed similarly as reported (see above, Scheme 1) [16].
![[1860-5397-16-213-i8]](/bjoc/content/inline/1860-5397-16-213-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Proposed reaction pathways starting from 10a in the presence of ACF and silane.
Scheme 8: Proposed reaction pathways starting from 10a in the presence of ACF and silane.
Overall, the reactivity study on 10a in the presence of ACF and HSiEt3 suggests that the formation of 1 described in the top part of the mechanism is favored when no silane is present in the reaction mixture or when only a small amount of silane is present (Scheme 8). In contrast, in neat silane, the bottom part of Scheme 8 is preferred, leading to the formation of 6.
Activation of 1,1,1,3,3-pentafluoropropane (HFC-245fa, 10c)
The reactivity of 1,1,1,3,3-pentafluoropropane (10c) at ACF was compared with the one of the isomer 1,1,1,2,3-pentafluoropropane (10a), again to elucidate conceivable reaction pathways and to understand potential similarities in their reactivity. In contrast to the findings for 10a, no conversion was observed without the use of HSiEt3 as a hydrogen source, indicating that for the activation of CHF2 groups, silane might be required.
When 10c was treated with 0.5 equivalents of HSiEt3 with respect to the substrate in the presence of ACF at 70 °C, the selective generation of the 1,3,3,3-tetrafluoropropenes (HFO-1234ze, 4a and 4b) was detected with an E:Z ratio of 10:1 and 43% conversion (Scheme 9). The transformation of 10c into 4a and 4b is remarkable since other catalytic conversions at chromia-based catalysts require elevated temperatures [11,19,59-62].
![[1860-5397-16-213-i9]](/bjoc/content/inline/1860-5397-16-213-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reactivity of 10c in the presence of ACF as the catalyst and 0.5 equivalents of HSiEt3 as a hydrogen source in C6D12.
Scheme 9: Reactivity of 10c in the presence of ACF as the catalyst and 0.5 equivalents of HSiEt3 as a hydroge...
The mechanisms for the C−F bond cleavage starting from 10c to yield the isomers 4 are similar to the ones proposed in the activation of 10a in the presence of silane (see above, Scheme 5). It is likely that silylium species are involved in the C–F activation steps at the ACF surface since HSiEt3 is needed to initiate any reactivity. On the one hand, an initial C–F bond activation at 10c by some silylium ion species will produce FSiEt3 and the corresponding carbenium species (Scheme 10). The latter can generate 4a and 4b together with H2, leading to the regeneration of the catalyst. When only 0.5 equivalents of silane are present, the reaction does not pursue further, as shown above (Scheme 9). On the other hand, as for 10a, an alternative mechanism can be proposed where the carbenium species is initially generated by an abstraction of a fluoride ion at the CHF2 group by the surface of the catalyst. Via HF elimination, 4a and 4b are produced, and the conversion is driven by the HF reaction with HSiEt3 to give FSiEt3.
![[1860-5397-16-213-i10]](/bjoc/content/inline/1860-5397-16-213-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Proposed catalytic cycles for the transformation of 10c in C6D12 and in the presence of 0.5 equivalents of silane and ACF as the catalyst.
Scheme 10: Proposed catalytic cycles for the transformation of 10c in C6D12 and in the presence of 0.5 equival...
When the amount of silane was increased to one equivalent, further reactivity was observed (Scheme 11). In C6D12, 6 was generated as the main compound, with traces of 5 and 14 (Scheme 11, top). When C6D6 was used as the solvent, 5 as the main product, 6 as a minor product, and 14 in traces were again generated, together with the Friedel–Crafts products CF2=CHCH2C6D5 (9) and CF3CH=CHC6D5 (8, Scheme 11, middle). In neat silane, 5 and 6 were detected in a ratio of 1:4 (Scheme 11, bottom).
![[1860-5397-16-213-i11]](/bjoc/content/inline/1860-5397-16-213-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Reactivity of 10c in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (top) or C6D6 (middle) as solvents or in neat silane (bottom).
Scheme 11: Reactivity of 10c in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (...
Notably, for all conversions (C6D6 and silane, C6D12 and silane, or in neat silane), monitoring of the reaction by 19F NMR spectroscopy led to the observation of the early generation of 4a and 4b after 24 hours reaction time at 70 °C. After 3 days at 70 °C, the transformation into the different products described above could be detected. Moreover, the continuous formation of FSiEt3 over time was observed for all reactions, which underlines the crucial role of the hydrogen source at each reaction step. As stated, it has been reported before that for the activation of 1,3,3,3-tetrafluoropropene (HFO-1234ze, 4) at ACF in neat HSiEt3 1,1-difluoropropene (5) and 1,1,1-trifluoropropane (6) are furnished (Scheme 1) [16]. In the presence of C6D6 and silane, the Friedel–Crafts products CF2=CHCH2C6D5 (9) and CF3CH=CHC6D5 (8) were observed, together with the hydroarylation product CF3CHDCH2C6D5 (7) [16]. To note, in neat silane, a distinct selectivity was detected, depending on the substrates used. Indeed, starting from 10c, the product ratio between 5 and 6 was 1:4, whereas it was reported to be 1:0.8 starting from 4a [16]. This observation might be the result of a hydrofluorination reaction from 5 to 6. Moreover, in neat silane, 14 was not detected, whereas when less silane and a solvent was present, this product was observed. Due to a large amount of silane present, a more considerable amount of the silylium species can be generated compared to when only one equivalent of silane is used. Therefore, the rate of the reaction from 10c to 6 might be increased in neat silane favoring two subsequent hydrodefluorination steps from 10c to yield 6.
As observed in the study on the reactivity of 10a, the variety of products for a larger amount of silane can be explained by several consecutive reactions, which include C–F bond activation steps (Scheme 12). Via hydrodefluorination, 10c can produce the intermediate 1,1,1,3-tetrafluoropropane (not observed). This intermediate could give 6 via a second hydrodefluorination. Alternatively, 1,1,1,3-tetrafluoropropane could also produce 14, which was shown to give 6 via the intermediate 5 by independent reactions. However, pathways to yield 6 are also required from the olefins 4, because they were detected as intermediates. This implies HF addition to 4 or 5. In the presence of benzene, 14 can also generate the Friedel–Crafts product 9; however, product 9 can also be generated from 4 by hydrodefluorination followed by a Friedel–Crafts reaction. The Friedel–Crafts product 8 can as well be formed from 10c directly, via a Friedel–Crafts reaction followed by HF elimination.
![[1860-5397-16-213-i12]](/bjoc/content/inline/1860-5397-16-213-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Proposed reaction pathways starting from 10c in the presence of ACF and silane.
Scheme 12: Proposed reaction pathways starting from 10c in the presence of ACF and silane.
Comparable to the reaction patterns of 10a, the reactivity study on 10c reveals that with a small amount of silane, the dehydrofluorination to 4 is favored, and with more silane, the conversions in Scheme 12 end up favorably in the formation of 6.
Activation of 1,1,1,2,2-pentafluoropropane (HFC-245fa, 10b)
As observed for 10c, the isomeric 1,1,1,2,2-pentafluoropropane (HFC-245fa, 10b) could also not be activated without the presence of HSiEt3 as a hydrogen source. The treatment of 10b in C6D12 in the presence of silane at 70 °C gave the allylic hydrodefluorination product 2 as well as 5 in a ratio of 1:1 with 38% conversion (Scheme 13, top). When benzene was used as a solvent together with silane, the Friedel–Crafts products 3 and 9 were observed in a ratio of 1:1, but the conversion reached only 18% (Scheme 13, middle). In neat silane, 2 and 5 were again observed, but this time the conversion did not exceed 10% (Scheme 13, bottom).
![[1860-5397-16-213-i13]](/bjoc/content/inline/1860-5397-16-213-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Reactivity of 10b in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (top) or C6D6 (middle) as solvents or in neat silane (bottom).
Scheme 13: Reactivity of 10b in the presence of ACF as the catalyst and HSiEt3 as a hydrogen source in C6D12 (...
As for the activation of 10c, it is plausible that surface silylium ion species are formed at ACF. Therefore, a reaction pathway can be suggested, starting with the initial generation of 1, FSiEt3, and H2. Subsequently, 2 can be generated via an allylic hydrodefluorination as it was observed in the study of tetrafluoropropenes at ACF [16]. Additionally, 5 can stem from several C–F bond activations at 10b, starting with a hydrodefluorination to generate the intermediate 13, which further undergoes an HF elimination, followed by an allylic hydrodefluorination to give 5. In the presence of benzene, at 1, a Friedel–Crafts reaction can generate 3, which further supports the formation as an intermediate of 1. Moreover, the intermediate 14 can also produce 9 via a Friedel–Crafts reaction, which again supports the pathway proposed to achieve 5.
Overall, for the reactivity of the pentafluoropropane isomer 10b, two different pathways seem to compete. The upper part of the reaction patterns in Scheme 14 leads to the formation of 2, and the bottom part provides pathways to 5. Thus, the hydrodefluorination step at 10b to form 13 seems to be more difficult than for the other isomers 10a and 10c.
![[1860-5397-16-213-i14]](/bjoc/content/inline/1860-5397-16-213-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Proposed reaction pathway starting from 10b in the presence of ACF and silane.
Scheme 14: Proposed reaction pathway starting from 10b in the presence of ACF and silane.
Conclusion
This study on the reactivity of pentafluoropropane isomers has revealed that ACF is a suitable catalyst for dehydrofluorination and hydrodefluorination reactions of polyfluorinated compounds under mild conditions. It was elaborated that the single C–F bond in 10a does not require the use of HSiEt3 as a hydrogen source to be activated via dehydrofluorination. However, when silane was introduced, further reactivity was observed, leading to the formation of subsequent defluorination products. In contrast, CF2, CHF2, and CF3 groups need the presence of a hydrogen source in order to promote the activation of at least one C–F bond by the formation of the thermodynamically stable Si–F bond. This observation is consistent with a decrease of the bond dissociation energies of C–F bonds from trifluoromethyl via difluoromethyl to monofluoromethyl groups [63]. Additionally, the C–F bond at the CHF2 group in 10c was easier to activate than the C–F bond in the CF3CF2 group in 10c, resulting in an order of reactivity of 10a > 10c > 10b. When only a small amount of silane was introduced for the reaction of 10a or 10c, the major products are due to dehydrofluorination, whereas in neat silane, formally hydrodefluorination products are generated mainly. By using C6D6, various Friedel–Crafts products can be further generated. For 10b, the conversions are very low, but dehydrofluorination and hydrodefluorination pathways compete with each other.
Mechanistically, the C–F bonds of the fluorinated substrates can be activated by Lewis-acidic sites at the ACF surface. In the presence of silane, it can be assumed that preferentially silylium surface species initiate the C–F bond cleavage. For both, the generated carbenium species show further reactivity to result in dehydrofluorination, hydrodefluorination, or Friedel–Crafts products. Notably, the conversion in neat silane was lower in the case of 10c and 10b, possibly because of a certain blocking of the acidic sites of ACF by silane. Note in that context that there are reports showing that silylium species can interact with more silane to generate larger entities [56,58,64].
Experimental
Material and methods
The reactions were carried out using Schlenk techniques as well as JYoung NMR tubes. The solvents were purchased from Eurisotop. C6D12 was dried over molecular sieves and purged with argon prior to use. C6D6 was dried with K-Solvona and distilled prior to use. Et3SiH (99%) was purchased from Sigma–Aldrich in a sure seal bottle and stored under argon. 1,1,1,2,3-Pentafluoropropane (HFC-245eb, 10a), 1,1,1,3,3-pentafluoropropane (HFC-245fa, 10c), and 1,1,1,2,2-pentafluoropropane (HFC-245cb, 10b) were gifted by Arkema and used without further purification. 3,3,3-Trifluoropropene (14, 99%), 1,1,1-trifluoropropane (6), and 1,1,1,2-tetrafluoropropane (13) were purchased from abcr and used without further purification. 1,1-Difluoropropene (5) was bought from Apollo Scientific and used without further purification. ACF was synthesized according to the literature and stored in a glove box. The number of active sites (1 mmol acidic sites/g of catalyst) was determined by temperature programmed desorption of ammonia (NH3-TPD) [34,65]. NMR spectra were recorded at room temperature using a Bruker DPX 300 spectrometer. A capillary of trifluorotoluene was employed as an external standard for quantification purposes. The 19F NMR spectra were referenced to PhCF3 (δ = −63.5 ppm) and the chemical shifts in 1H NMR were referenced to residual C6D5H (δ = 7.16 ppm) or C6D11H (δ = 1.38 ppm).
Procedure for reactions with gaseous substrates
A JYoung NMR tube was loaded with 25 mg of ACF inside a glovebox. In experiments involving a solvent (C6D6 or C6D12), 0.4 mL of the solvent was added under Schlenk conditions, together with the corresponding amount of HSiEt3. In the reactions without solvent, 0.5 mL of HSiEt3 was added using Schlenk techniques to the JYoung NMR tube loaded with ACF. The gases were then condensed using a small glass bulb filled with 0.5 atm of the corresponding gas (0.1 mmol). The reactions were monitored by 1H and 19F NMR spectroscopy. The tubes were kept at 70 °C for 7 days. PhCF3 was used as an external standard in a closed capillary to calculate the conversion based on the consumed substrate by the integration of the 19F NMR spectra.
Supporting Information
| Supporting Information File 1: Analytical data and copies of spectra. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Velders, G. J. M.; Andersen, S. O.; Daniel, J. S.; Fahey, D. W.; McFarland, M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4814–4819. doi:10.1073/pnas.0610328104
Return to citation in text: [1] -
Ko, M.; Shia, R.-L.; Sze, N.-D.; Magid, H.; Bray, R. G. J. Geophys. Res.: Atmos. 1999, 104, 8173–8181. doi:10.1029/1998jd100097
Return to citation in text: [1] -
Bolaji, B. O.; Huan, Z. Renewable Sustainable Energy Rev. 2013, 18, 49–54. doi:10.1016/j.rser.2012.10.008
Return to citation in text: [1] -
United Nations Environment Programme; Handbook for the Montreal Protocol on Substances That Deplete the Ozone Layer; 2018.
Return to citation in text: [1] -
United Nations Environment Programme; The Montreal Protocol on Substances That Deplete the Ozone Layer; 2000. doi:10.1163/15718069620847781
Return to citation in text: [1] -
United Nations Environment Programme; Production and Consumption of Ozone Depleting Substances under the Montreal Protocol; Nairobi, 2005.
Return to citation in text: [1] -
United Nations Environment Programme; Amendment to the Montreal Protocol on Substances That Deplete the Ozone Layer; 2016.
Return to citation in text: [1] -
United Nations Environment Programme; Ratification of the Kigali Amendment; 2017.
Return to citation in text: [1] -
United Nations Environment Programme; The Kigali Amendment to the Montreal Protocol: HFC Phase-Down; 2016.
Return to citation in text: [1] -
Velders, G. J. M.; Ravishankara, A. R.; Miller, M. K.; Molina, M. J.; Alcamo, J.; Daniel, J. S.; Fahey, D. W.; Montzka, S. A.; Reimann, S. Science 2012, 335, 922–923. doi:10.1126/science.1216414
Return to citation in text: [1] -
Wang, H.; Tung, H. S. Process for the Production of HFO Trans-1234ze from HFC-245fa. U.S. Patent US8,067,650 B2, July 10, 2011.
Return to citation in text: [1] [2] [3] [4] -
Light, B. A.; Phillips, S. D.; Fleming, K. M.; Ferguson, S. A.; Ma, J. J.; Bortz, C. L.; Van Der Puy, M.; Merkel, D. C.; Tung, H. S.; Mukhopadhyay, S. Method for Producing Fluorinated Organic Compounds. U.S. Patent US9102579 B2, Jan 3, 2007.
Return to citation in text: [1] [2] -
Righetti, G.; Zilio, C.; Longo, G. A. Int. J. Refrig. 2015, 54, 1–9. doi:10.1016/j.ijrefrig.2015.02.010
Return to citation in text: [1] [2] -
Fang, X.-X.; Wang, Y.; Jia, W.-Z.; Song, J.-D.; Wang, Y.-J.; Luo, M.-F.; Lu, J.-Q. Appl. Catal., A 2019, 576, 39–46. doi:10.1016/j.apcata.2019.02.035
Return to citation in text: [1] [2] -
Van Der Puy, M. Process for the Preparation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf). U.S. Patent US8,071,826 B2, Dec 6, 2011.
Return to citation in text: [1] [2] [3] -
Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] -
Lim, S.; Kim, M. S.; Choi, J.-W.; Kim, H.; Ahn, B. S.; Lee, S. D.; Lee, H.; Kim, C. S.; Suh, D. J.; Ha, J.-M.; Song, K. H. Catal. Today 2017, 293–294, 42–48. doi:10.1016/j.cattod.2016.11.017
Return to citation in text: [1] [2] -
Wang, F.; Zhang, W.; Liang, Y.; Wang, Y.; Lu, J.; Luo, M. Chem. Res. Chin. Univ. 2015, 31, 1003–1006. doi:10.1007/s40242-015-5190-3
Return to citation in text: [1] -
Mao, W.; Bai, Y.; Jia, Z.; Yang, Z.; Hao, Z.; Lu, J. Appl. Catal., A 2018, 564, 147–156. doi:10.1016/j.apcata.2018.07.028
Return to citation in text: [1] [2] -
Shen, Q.; Huang, Y.-G.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y. J. Fluorine Chem. 2015, 179, 14–22. doi:10.1016/j.jfluchem.2015.07.007
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/c6cs00351f
Return to citation in text: [1] -
Gilles, P. W.; Margrave, J. L. J. Chem. Phys. 1953, 21, 381–382. doi:10.1063/1.1698915
Return to citation in text: [1] -
Mazurek, U.; Schwarz, H. Chem. Commun. 2003, 1321–1326. doi:10.1039/b211850e
Return to citation in text: [1] -
Braun, T.; Wehmeier, F. Eur. J. Inorg. Chem. 2011, 613–625. doi:10.1002/ejic.201001184
Return to citation in text: [1] -
Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] [2] -
Krahl, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 773–796. doi:10.1039/c6cy02369j
Return to citation in text: [1] [2] -
Stahl, T.; Klare, H. F. T.; Oestreich, M. ACS Catal. 2013, 3, 1578–1587. doi:10.1021/cs4003244
Return to citation in text: [1] -
Fuchibe, K.; Hatta, H.; Oh, K.; Oki, R.; Ichikawa, J. Angew. Chem., Int. Ed. 2017, 56, 5890–5893. doi:10.1002/anie.201701985
Return to citation in text: [1] -
Yagupolskii, Yu. L.; Pavlenko, N. V.; Shelyazhenko, S. V.; Filatov, A. A.; Kremlev, M. M.; Mushta, A. I.; Gerus, I. I.; Peng, S.; Petrov, V. A.; Nappa, M. J. Fluorine Chem. 2015, 179, 134–141. doi:10.1016/j.jfluchem.2015.08.001
Return to citation in text: [1] -
Teinz, K.; Wuttke, S.; Börno, F.; Eicher, J.; Kemnitz, E. J. Catal. 2011, 282, 175–182. doi:10.1016/j.jcat.2011.06.013
Return to citation in text: [1] -
Teinz, K.; Manuel, S. R.; Chen, B. B.; Pigamo, A.; Doucet, N.; Kemnitz, E. Appl. Catal., B 2015, 165, 200–208. doi:10.1016/j.apcatb.2014.09.076
Return to citation in text: [1] [2] [3] -
Prechtl, M. H. G.; Teltewskoi, M.; Dimitrov, A.; Kemnitz, E.; Braun, T. Chem. – Eur. J. 2011, 17, 14385–14388. doi:10.1002/chem.201102853
Return to citation in text: [1] [2] -
Calvo, B.; Braun, T.; Kemnitz, E. ChemCatChem 2018, 10, 403–406. doi:10.1002/cctc.201701327
Return to citation in text: [1] -
Calvo, B.; Wuttke, J.; Braun, T.; Kemnitz, E. ChemCatChem 2016, 8, 1945–1950. doi:10.1002/cctc.201600257
Return to citation in text: [1] -
Siwek, A. K.; Ahrens, M.; Feist, M.; Braun, T.; Kemnitz, E. ChemCatChem 2017, 9, 839–845. doi:10.1002/cctc.201601238
Return to citation in text: [1] -
Meißner, G.; Feist, M.; Braun, T.; Kemnitz, E. J. Organomet. Chem. 2017, 847, 234–241. doi:10.1016/j.jorganchem.2017.04.030
Return to citation in text: [1] [2] -
Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Krahl, T.; Kemnitz, E. J. Fluorine Chem. 2006, 127, 663–678. doi:10.1016/j.jfluchem.2006.02.015
Return to citation in text: [1] [2] -
Krahl, T.; Stösser, R.; Kemnitz, E.; Scholz, G.; Feist, M.; Silly, G.; Buzaré, J.-Y. Inorg. Chem. 2003, 42, 6474–6483. doi:10.1021/ic034106h
Return to citation in text: [1] [2] [3] -
Krespan, C. G. Process for Production of Polyfluoroolefins. U.S. Pat. Appl. US 5,162,594 A, Nov 11, 1992.
Return to citation in text: [1] -
Calvo, B.; Marshall, C. P.; Krahl, T.; Kröhnert, J.; Trunschke, A.; Scholz, G.; Braun, T.; Kemnitz, E. Dalton Trans. 2018, 47, 16461–16473. doi:10.1039/c8dt03279c
Return to citation in text: [1] -
Petrov, V. A.; Krespan, C. G.; Smart, B. E. J. Fluorine Chem. 1998, 89, 125–130. doi:10.1016/s0022-1139(98)00098-0
Return to citation in text: [1] -
Petrov, V. A.; Krespan, C. G.; Smart, B. E. J. Fluorine Chem. 1996, 77, 139–142. doi:10.1016/0022-1139(96)03391-x
Return to citation in text: [1] -
Petrov, V. A.; Krespan, C. G. J. Fluorine Chem. 2000, 102, 199–204. doi:10.1016/s0022-1139(99)00279-1
Return to citation in text: [1] -
Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. doi:10.1002/anie.201300608
Return to citation in text: [1] [2] [3] [4] -
Luo, Y. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007.
Return to citation in text: [1] -
Kuehnel, M. F.; Lentz, D.; Braun, T. Angew. Chem., Int. Ed. 2013, 52, 3328–3348. doi:10.1002/anie.201205260
Return to citation in text: [1] -
Kervarec, M.-C.; Marshall, C. P.; Braun, T.; Kemnitz, E. J. Fluorine Chem. 2019, 221, 61–65. doi:10.1016/j.jfluchem.2019.04.001
Return to citation in text: [1] [2] [3] -
Xie, Z.; Fan, J.; Cheng, Y.; Jin, L.; Hu, G.; Lu, J.; Luo, M. Ind. Eng. Chem. Res. 2013, 52, 3295–3299. doi:10.1021/ie303067d
Return to citation in text: [1] -
Wang, H.; Tung, H. S.; Singh, R. R.; Shankland, I. Process for Producing 2,3,3,3-Tetrafluoropropene. U.S. Patent US8,766,020 B2, July 1, 2014.
Return to citation in text: [1] -
Braun, T.; Wehmeier, F.; Altenhöner, K. Angew. Chem., Int. Ed. 2007, 46, 5321–5324. doi:10.1002/anie.200700711
Return to citation in text: [1] -
Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188–1190. doi:10.1126/science.1159979
Return to citation in text: [1] -
Douvris, C.; Nagaraja, C. M.; Chen, C.-H.; Foxman, B. M.; Ozerov, O. V. J. Am. Chem. Soc. 2010, 132, 4946–4953. doi:10.1021/ja100605m
Return to citation in text: [1] -
Scott, V. J.; Çelenligil-Çetin, R.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852–2853. doi:10.1021/ja0426138
Return to citation in text: [1] [2] -
Panisch, R.; Bolte, M.; Müller, T. J. Am. Chem. Soc. 2006, 128, 9676–9682. doi:10.1021/ja061800y
Return to citation in text: [1] -
Talavera, M.; Meißner, G.; Rachor, S. G.; Braun, T. Chem. Commun. 2020, 56, 4452–4455. doi:10.1039/d0cc01420f
Return to citation in text: [1] [2] -
Song, J.-D.; Song, T.-Y.; Zhang, T.-T.; Wang, Y.; Luo, M.-F.; Lu, J.-Q. J. Catal. 2018, 364, 271–281. doi:10.1016/j.jcat.2018.04.014
Return to citation in text: [1] -
Wang, F.; Liang, Y.; Zhang, W.-X.; Wang, Y.-J.; Lu, J.-Q.; Meng-Fei, L. Indian J. Chem. 2015, 54, 1192–1197.
Return to citation in text: [1] -
Tung, H. S.; Johnson, R. C.; Merkel, D. C. Process for the Manufacture of 1,3,3,3-Tetrafluoropropene. U.S. Patent US7,592,494 B2, Sept 22, 2009.
Return to citation in text: [1] -
Pen, S.; Nappa, M. J. Dehydrofluorination of 245fa to 1234ze. U.S. Patent US9,302,962 B2, April 5, 2016.
Return to citation in text: [1] -
Burdeniuc, J.; Jedicka, B.; Crabtree, R. H. Chem. Ber./Recl. 1997, 130, 145–154. doi:10.1002/cber.19971300203
Return to citation in text: [1] -
Müller, T. Silylium Ions. In Functional Molecular Silicon Compounds I. Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Cham, Switzerland, 2013; Vol. 155, pp 107–162. doi:10.1007/430_2013_132
Return to citation in text: [1] -
Murthy, J. K.; Gross, U.; Rüdiger, S.; Rao, V. V.; Kumar, V. V.; Wander, A.; Bailey, C. L.; Harrison, N. M.; Kemnitz, E. J. Phys. Chem. B 2006, 110, 8314–8319. doi:10.1021/jp0601419
Return to citation in text: [1]
| 54. | Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188–1190. doi:10.1126/science.1159979 |
| 55. | Douvris, C.; Nagaraja, C. M.; Chen, C.-H.; Foxman, B. M.; Ozerov, O. V. J. Am. Chem. Soc. 2010, 132, 4946–4953. doi:10.1021/ja100605m |
| 56. | Scott, V. J.; Çelenligil-Çetin, R.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852–2853. doi:10.1021/ja0426138 |
| 57. | Panisch, R.; Bolte, M.; Müller, T. J. Am. Chem. Soc. 2006, 128, 9676–9682. doi:10.1021/ja061800y |
| 58. | Talavera, M.; Meißner, G.; Rachor, S. G.; Braun, T. Chem. Commun. 2020, 56, 4452–4455. doi:10.1039/d0cc01420f |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 50. | Kervarec, M.-C.; Marshall, C. P.; Braun, T.; Kemnitz, E. J. Fluorine Chem. 2019, 221, 61–65. doi:10.1016/j.jfluchem.2019.04.001 |
| 1. | Velders, G. J. M.; Andersen, S. O.; Daniel, J. S.; Fahey, D. W.; McFarland, M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4814–4819. doi:10.1073/pnas.0610328104 |
| 2. | Ko, M.; Shia, R.-L.; Sze, N.-D.; Magid, H.; Bray, R. G. J. Geophys. Res.: Atmos. 1999, 104, 8173–8181. doi:10.1029/1998jd100097 |
| 3. | Bolaji, B. O.; Huan, Z. Renewable Sustainable Energy Rev. 2013, 18, 49–54. doi:10.1016/j.rser.2012.10.008 |
| 11. | Wang, H.; Tung, H. S. Process for the Production of HFO Trans-1234ze from HFC-245fa. U.S. Patent US8,067,650 B2, July 10, 2011. |
| 14. | Fang, X.-X.; Wang, Y.; Jia, W.-Z.; Song, J.-D.; Wang, Y.-J.; Luo, M.-F.; Lu, J.-Q. Appl. Catal., A 2019, 576, 39–46. doi:10.1016/j.apcata.2019.02.035 |
| 15. | Van Der Puy, M. Process for the Preparation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf). U.S. Patent US8,071,826 B2, Dec 6, 2011. |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 11. | Wang, H.; Tung, H. S. Process for the Production of HFO Trans-1234ze from HFC-245fa. U.S. Patent US8,067,650 B2, July 10, 2011. |
| 12. | Light, B. A.; Phillips, S. D.; Fleming, K. M.; Ferguson, S. A.; Ma, J. J.; Bortz, C. L.; Van Der Puy, M.; Merkel, D. C.; Tung, H. S.; Mukhopadhyay, S. Method for Producing Fluorinated Organic Compounds. U.S. Patent US9102579 B2, Jan 3, 2007. |
| 13. | Righetti, G.; Zilio, C.; Longo, G. A. Int. J. Refrig. 2015, 54, 1–9. doi:10.1016/j.ijrefrig.2015.02.010 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 7. | United Nations Environment Programme; Amendment to the Montreal Protocol on Substances That Deplete the Ozone Layer; 2016. |
| 8. | United Nations Environment Programme; Ratification of the Kigali Amendment; 2017. |
| 9. | United Nations Environment Programme; The Kigali Amendment to the Montreal Protocol: HFC Phase-Down; 2016. |
| 10. | Velders, G. J. M.; Ravishankara, A. R.; Miller, M. K.; Molina, M. J.; Alcamo, J.; Daniel, J. S.; Fahey, D. W.; Montzka, S. A.; Reimann, S. Science 2012, 335, 922–923. doi:10.1126/science.1216414 |
| 47. | Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. doi:10.1002/anie.201300608 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 4. | United Nations Environment Programme; Handbook for the Montreal Protocol on Substances That Deplete the Ozone Layer; 2018. |
| 5. | United Nations Environment Programme; The Montreal Protocol on Substances That Deplete the Ozone Layer; 2000. doi:10.1163/15718069620847781 |
| 6. | United Nations Environment Programme; Production and Consumption of Ozone Depleting Substances under the Montreal Protocol; Nairobi, 2005. |
| 27. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 48. | Luo, Y. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. |
| 49. | Kuehnel, M. F.; Lentz, D.; Braun, T. Angew. Chem., Int. Ed. 2013, 52, 3328–3348. doi:10.1002/anie.201205260 |
| 11. | Wang, H.; Tung, H. S. Process for the Production of HFO Trans-1234ze from HFC-245fa. U.S. Patent US8,067,650 B2, July 10, 2011. |
| 19. | Mao, W.; Bai, Y.; Jia, Z.; Yang, Z.; Hao, Z.; Lu, J. Appl. Catal., A 2018, 564, 147–156. doi:10.1016/j.apcata.2018.07.028 |
| 59. | Song, J.-D.; Song, T.-Y.; Zhang, T.-T.; Wang, Y.; Luo, M.-F.; Lu, J.-Q. J. Catal. 2018, 364, 271–281. doi:10.1016/j.jcat.2018.04.014 |
| 60. | Wang, F.; Liang, Y.; Zhang, W.-X.; Wang, Y.-J.; Lu, J.-Q.; Meng-Fei, L. Indian J. Chem. 2015, 54, 1192–1197. |
| 61. | Tung, H. S.; Johnson, R. C.; Merkel, D. C. Process for the Manufacture of 1,3,3,3-Tetrafluoropropene. U.S. Patent US7,592,494 B2, Sept 22, 2009. |
| 62. | Pen, S.; Nappa, M. J. Dehydrofluorination of 245fa to 1234ze. U.S. Patent US9,302,962 B2, April 5, 2016. |
| 20. | Shen, Q.; Huang, Y.-G.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y. J. Fluorine Chem. 2015, 179, 14–22. doi:10.1016/j.jfluchem.2015.07.007 |
| 21. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 22. | Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/c6cs00351f |
| 23. | Gilles, P. W.; Margrave, J. L. J. Chem. Phys. 1953, 21, 381–382. doi:10.1063/1.1698915 |
| 24. | Mazurek, U.; Schwarz, H. Chem. Commun. 2003, 1321–1326. doi:10.1039/b211850e |
| 25. | Braun, T.; Wehmeier, F. Eur. J. Inorg. Chem. 2011, 613–625. doi:10.1002/ejic.201001184 |
| 26. | Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c |
| 27. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 28. | Krahl, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 773–796. doi:10.1039/c6cy02369j |
| 32. | Teinz, K.; Wuttke, S.; Börno, F.; Eicher, J.; Kemnitz, E. J. Catal. 2011, 282, 175–182. doi:10.1016/j.jcat.2011.06.013 |
| 33. | Teinz, K.; Manuel, S. R.; Chen, B. B.; Pigamo, A.; Doucet, N.; Kemnitz, E. Appl. Catal., B 2015, 165, 200–208. doi:10.1016/j.apcatb.2014.09.076 |
| 34. | Prechtl, M. H. G.; Teltewskoi, M.; Dimitrov, A.; Kemnitz, E.; Braun, T. Chem. – Eur. J. 2011, 17, 14385–14388. doi:10.1002/chem.201102853 |
| 35. | Calvo, B.; Braun, T.; Kemnitz, E. ChemCatChem 2018, 10, 403–406. doi:10.1002/cctc.201701327 |
| 36. | Calvo, B.; Wuttke, J.; Braun, T.; Kemnitz, E. ChemCatChem 2016, 8, 1945–1950. doi:10.1002/cctc.201600257 |
| 37. | Siwek, A. K.; Ahrens, M.; Feist, M.; Braun, T.; Kemnitz, E. ChemCatChem 2017, 9, 839–845. doi:10.1002/cctc.201601238 |
| 38. | Meißner, G.; Feist, M.; Braun, T.; Kemnitz, E. J. Organomet. Chem. 2017, 847, 234–241. doi:10.1016/j.jorganchem.2017.04.030 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 41. | Krahl, T.; Stösser, R.; Kemnitz, E.; Scholz, G.; Feist, M.; Silly, G.; Buzaré, J.-Y. Inorg. Chem. 2003, 42, 6474–6483. doi:10.1021/ic034106h |
| 19. | Mao, W.; Bai, Y.; Jia, Z.; Yang, Z.; Hao, Z.; Lu, J. Appl. Catal., A 2018, 564, 147–156. doi:10.1016/j.apcata.2018.07.028 |
| 40. | Krahl, T.; Kemnitz, E. J. Fluorine Chem. 2006, 127, 663–678. doi:10.1016/j.jfluchem.2006.02.015 |
| 41. | Krahl, T.; Stösser, R.; Kemnitz, E.; Scholz, G.; Feist, M.; Silly, G.; Buzaré, J.-Y. Inorg. Chem. 2003, 42, 6474–6483. doi:10.1021/ic034106h |
| 42. | Krespan, C. G. Process for Production of Polyfluoroolefins. U.S. Pat. Appl. US 5,162,594 A, Nov 11, 1992. |
| 43. | Calvo, B.; Marshall, C. P.; Krahl, T.; Kröhnert, J.; Trunschke, A.; Scholz, G.; Braun, T.; Kemnitz, E. Dalton Trans. 2018, 47, 16461–16473. doi:10.1039/c8dt03279c |
| 44. | Petrov, V. A.; Krespan, C. G.; Smart, B. E. J. Fluorine Chem. 1998, 89, 125–130. doi:10.1016/s0022-1139(98)00098-0 |
| 45. | Petrov, V. A.; Krespan, C. G.; Smart, B. E. J. Fluorine Chem. 1996, 77, 139–142. doi:10.1016/0022-1139(96)03391-x |
| 46. | Petrov, V. A.; Krespan, C. G. J. Fluorine Chem. 2000, 102, 199–204. doi:10.1016/s0022-1139(99)00279-1 |
| 40. | Krahl, T.; Kemnitz, E. J. Fluorine Chem. 2006, 127, 663–678. doi:10.1016/j.jfluchem.2006.02.015 |
| 41. | Krahl, T.; Stösser, R.; Kemnitz, E.; Scholz, G.; Feist, M.; Silly, G.; Buzaré, J.-Y. Inorg. Chem. 2003, 42, 6474–6483. doi:10.1021/ic034106h |
| 11. | Wang, H.; Tung, H. S. Process for the Production of HFO Trans-1234ze from HFC-245fa. U.S. Patent US8,067,650 B2, July 10, 2011. |
| 14. | Fang, X.-X.; Wang, Y.; Jia, W.-Z.; Song, J.-D.; Wang, Y.-J.; Luo, M.-F.; Lu, J.-Q. Appl. Catal., A 2019, 576, 39–46. doi:10.1016/j.apcata.2019.02.035 |
| 15. | Van Der Puy, M. Process for the Preparation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf). U.S. Patent US8,071,826 B2, Dec 6, 2011. |
| 17. | Lim, S.; Kim, M. S.; Choi, J.-W.; Kim, H.; Ahn, B. S.; Lee, S. D.; Lee, H.; Kim, C. S.; Suh, D. J.; Ha, J.-M.; Song, K. H. Catal. Today 2017, 293–294, 42–48. doi:10.1016/j.cattod.2016.11.017 |
| 18. | Wang, F.; Zhang, W.; Liang, Y.; Wang, Y.; Lu, J.; Luo, M. Chem. Res. Chin. Univ. 2015, 31, 1003–1006. doi:10.1007/s40242-015-5190-3 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 12. | Light, B. A.; Phillips, S. D.; Fleming, K. M.; Ferguson, S. A.; Ma, J. J.; Bortz, C. L.; Van Der Puy, M.; Merkel, D. C.; Tung, H. S.; Mukhopadhyay, S. Method for Producing Fluorinated Organic Compounds. U.S. Patent US9102579 B2, Jan 3, 2007. |
| 13. | Righetti, G.; Zilio, C.; Longo, G. A. Int. J. Refrig. 2015, 54, 1–9. doi:10.1016/j.ijrefrig.2015.02.010 |
| 15. | Van Der Puy, M. Process for the Preparation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf). U.S. Patent US8,071,826 B2, Dec 6, 2011. |
| 28. | Krahl, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 773–796. doi:10.1039/c6cy02369j |
| 29. | Stahl, T.; Klare, H. F. T.; Oestreich, M. ACS Catal. 2013, 3, 1578–1587. doi:10.1021/cs4003244 |
| 30. | Fuchibe, K.; Hatta, H.; Oh, K.; Oki, R.; Ichikawa, J. Angew. Chem., Int. Ed. 2017, 56, 5890–5893. doi:10.1002/anie.201701985 |
| 31. | Yagupolskii, Yu. L.; Pavlenko, N. V.; Shelyazhenko, S. V.; Filatov, A. A.; Kremlev, M. M.; Mushta, A. I.; Gerus, I. I.; Peng, S.; Petrov, V. A.; Nappa, M. J. Fluorine Chem. 2015, 179, 134–141. doi:10.1016/j.jfluchem.2015.08.001 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 33. | Teinz, K.; Manuel, S. R.; Chen, B. B.; Pigamo, A.; Doucet, N.; Kemnitz, E. Appl. Catal., B 2015, 165, 200–208. doi:10.1016/j.apcatb.2014.09.076 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 50. | Kervarec, M.-C.; Marshall, C. P.; Braun, T.; Kemnitz, E. J. Fluorine Chem. 2019, 221, 61–65. doi:10.1016/j.jfluchem.2019.04.001 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 63. | Burdeniuc, J.; Jedicka, B.; Crabtree, R. H. Chem. Ber./Recl. 1997, 130, 145–154. doi:10.1002/cber.19971300203 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 47. | Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. doi:10.1002/anie.201300608 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 47. | Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. doi:10.1002/anie.201300608 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 50. | Kervarec, M.-C.; Marshall, C. P.; Braun, T.; Kemnitz, E. J. Fluorine Chem. 2019, 221, 61–65. doi:10.1016/j.jfluchem.2019.04.001 |
| 53. | Braun, T.; Wehmeier, F.; Altenhöner, K. Angew. Chem., Int. Ed. 2007, 46, 5321–5324. doi:10.1002/anie.200700711 |
| 16. | Meißner, G.; Kretschmar, K.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. doi:10.1002/anie.201707759 |
| 39. | Meißner, G.; Dirican, D.; Jäger, C.; Braun, T.; Kemnitz, E. Catal. Sci. Technol. 2017, 7, 3348–3354. doi:10.1039/c7cy00845g |
| 47. | Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. doi:10.1002/anie.201300608 |
| 17. | Lim, S.; Kim, M. S.; Choi, J.-W.; Kim, H.; Ahn, B. S.; Lee, S. D.; Lee, H.; Kim, C. S.; Suh, D. J.; Ha, J.-M.; Song, K. H. Catal. Today 2017, 293–294, 42–48. doi:10.1016/j.cattod.2016.11.017 |
| 51. | Xie, Z.; Fan, J.; Cheng, Y.; Jin, L.; Hu, G.; Lu, J.; Luo, M. Ind. Eng. Chem. Res. 2013, 52, 3295–3299. doi:10.1021/ie303067d |
| 52. | Wang, H.; Tung, H. S.; Singh, R. R.; Shankland, I. Process for Producing 2,3,3,3-Tetrafluoropropene. U.S. Patent US8,766,020 B2, July 1, 2014. |
| 33. | Teinz, K.; Manuel, S. R.; Chen, B. B.; Pigamo, A.; Doucet, N.; Kemnitz, E. Appl. Catal., B 2015, 165, 200–208. doi:10.1016/j.apcatb.2014.09.076 |
| 56. | Scott, V. J.; Çelenligil-Çetin, R.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852–2853. doi:10.1021/ja0426138 |
| 58. | Talavera, M.; Meißner, G.; Rachor, S. G.; Braun, T. Chem. Commun. 2020, 56, 4452–4455. doi:10.1039/d0cc01420f |
| 64. | Müller, T. Silylium Ions. In Functional Molecular Silicon Compounds I. Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Cham, Switzerland, 2013; Vol. 155, pp 107–162. doi:10.1007/430_2013_132 |
| 38. | Meißner, G.; Feist, M.; Braun, T.; Kemnitz, E. J. Organomet. Chem. 2017, 847, 234–241. doi:10.1016/j.jorganchem.2017.04.030 |
| 34. | Prechtl, M. H. G.; Teltewskoi, M.; Dimitrov, A.; Kemnitz, E.; Braun, T. Chem. – Eur. J. 2011, 17, 14385–14388. doi:10.1002/chem.201102853 |
| 65. | Murthy, J. K.; Gross, U.; Rüdiger, S.; Rao, V. V.; Kumar, V. V.; Wander, A.; Bailey, C. L.; Harrison, N. M.; Kemnitz, E. J. Phys. Chem. B 2006, 110, 8314–8319. doi:10.1021/jp0601419 |
© 2020 Kervarec et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)