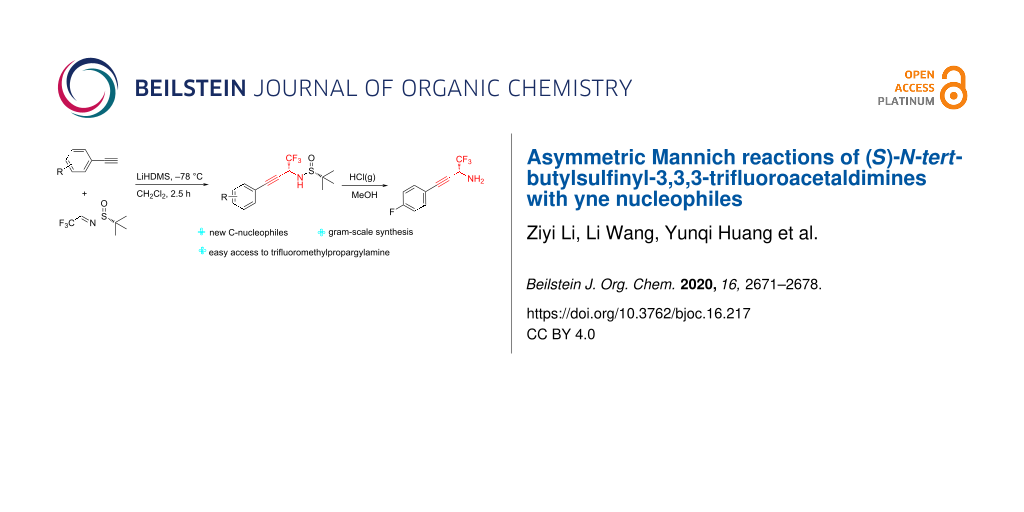
Abstract
In the present work, arylethynes were studied as new C-nucleophiles in the asymmetric Mannich addition reactions with (S)-N-tert-butylsulfinyl-3,3,3-trifluoroacetaldimine. The reactions were conducted under operationally convenient conditions affording the corresponding Mannich adducts with up to 87% yield and 70:30 diastereoselectivity. The isomeric products can be separated using regular column chromatography to afford diastereomerically pure compounds. The purified Mannich addition products were deprotected to give the target enantiomerically pure trifluoromethylpropargylamines. A mechanistic rationale for the observed stereochemical outcome is discussed.

Graphical Abstract
Introduction
In recent years, substitution of hydrogen by fluorine atoms or fluorine-containing groups usually provides unexpected biological and physicochemical properties, which thus has become an established approach for the development of pharmaceuticals [1-9], agrochemicals [10-14], and advanced materials [15-19]. On the other hand, chiral propargylamine represents a very important type of organic intermediates, which has been successfully used in the synthesis of natural products and biologically relevant heterocyclic compounds [20-24]. Thus, fluorinated propargylamine, in particular, chiral trifluoromethylpropargylamine, should be considered of great research interest due to the apparently advantageous pharmaceutical profile of CF3-containing drugs [25,26]. For example, DPC 961 contains the trifluoromethylpropargylamine moiety and has been developed as the inhibitor against non-nucleoside reverse transcriptase for the treatment of human immunodeficiency virus [27] (Figure 1).
![[1860-5397-16-217-1]](/bjoc/content/figures/1860-5397-16-217-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Anti-HIV compound containing a trifluoromethylpropargylamine moiety.
Figure 1: Anti-HIV compound containing a trifluoromethylpropargylamine moiety.
Thus, the development of synthetic methods for the preparation of these compounds, featuring trifluoromethylpropargylamine is of general research interest [28-32]. The asymmetric Ru/(R)-CPA-catalyzed chemoselectivie biomimetic reduction [33] and the organocatalytic transfer hydrogenation [34] of fluorinated alkynylketimines have been developed for the synthesis of fluorinated propargylamines in good yields and high enantioselectivities by the groups of Zhou and Peng, respectively (Scheme 1a). It should be mentioned that the Qing group also reported a method for the synthesis of α-trifluoromethylated α-propargylamines via a Ti-promoted addition reaction between acetylide and chiral CF3-ketimines (Scheme 1b) [35]. Based on our experience in the preparation of trifluoromethylated amino compounds [36-40] and the chemistry of N-tert-butylsulfinyl-3,3,3-trifluoroacetaldimine (1) [41-54], we noticed that the reactions of chiral imine 1 with yne nucleophiles have never been reported thus far. Accordingly, intrigued by this methodological deficiency, we decided to dedicate a special research project to this objective; herein, we report Mannich reactions between yne nucleophiles and aldimine 1 (Scheme 1c). The results reported in this work expand our knowledge of the reactivity of imines and the origins of stereocontrol, as well as provide synthetic access to a series of trifluoromethylpropargylamine of high biological interest.
![[1860-5397-16-217-i1]](/bjoc/content/inline/1860-5397-16-217-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Literature-known methods (a and b) and the here reported (c) approach for the synthesis of α-trifluoromethylated α-propargylamines.
Scheme 1: Literature-known methods (a and b) and the here reported (c) approach for the synthesis of α-triflu...
Results and Discussion
Drawing from our previous studies on the chemistry of trifluoromethylated sulfinylimine 1 [41-54], the initial reaction conditions focused on the use of 1.3 equiv of 1-ethynyl-4-methoxybenzene (2a) in the presence of n-BuLi with CH2Cl2 as a solvent at −78 °C. The desired product 3a was obtained after 2.5 hours in 46% yield as a mixture of two diastereomers (55:45 dr, Table 1, entry 1). Fortunately, the diastereomeric products can be quite easily separated by regular column chromatography using hexane/EtOAc (4:1, v/v) as an eluent. Further optimization of the reaction conditions was then carried out to improve both the yield and diastereoselectivity. First, a series of pilot experiments in dichloromethane were performed to scan the base in the reaction. When using LDA and LiHMDS instead of n-BuLi, the yields were noticeably increased to 83% and 87%, respectively (Table 1, entries 2 and 3). In particular, the use of LiHMDS resulted in improved diastereoselectivity (69:31 dr, Table 1, entry 3). On the other hand, the solvent was found to show a significant effect on this transformation (Table 1, entries 4–6) and the results indicated dichloromethane was the best choice. Variation of the loading amount of 1-ethynyl-4-methoxybenzene (2a) did not provide any improvement on the chemical yield and the stereochemical outcome (Table 1, entries 7 and 8). We also observed a noticeable temperature effect on the reaction yield (Table 1, entries 9 and 10) and −78 °C was proved to be the best choice. Screening of the reaction time revealed that 2.5 hours were necessary for the completion of this transformation (Table 1, entries 11 and 12). Finally, we found that the use of Lewis acids BF3·Et2O and Ti(OiPr)4 as additives for this reaction was unsuccessful and the same level of chemical yield and diastereoselectivity was observed (Table 1, entries 13 and 14).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-217-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Base | Solvent | 2a (equiv) | T (°C) | Time (h) | Yieldb (%) | drc |
| 1 | n-BuLi | CH2Cl2 | 1.3 | −78 | 2.5 | 46 | 55:45 |
| 2 | LDA | CH2Cl2 | 1.3 | −78 | 2.5 | 83 | 66:34 |
| 3 | LiHMDS | CH2Cl2 | 1.3 | −78 | 2.5 | 87 | 69:31 |
| 4 | LiHMDS | THF | 1.3 | −78 | 2.5 | 57 | 56:44 |
| 5 | LiHMDS | CHCl3 | 1.3 | −60 | 2.5 | trace | – |
| 6 | LiHMDS | PhCH3 | 1.3 | −78 | 2.5 | 45 | 63:37 |
| 7 | LiHMDS | CH2Cl2 | 1.1 | −78 | 2.5 | 85 | 66:34 |
| 8 | LiHMDS | CH2Cl2 | 1.6 | −78 | 2.5 | 79 | 68:32 |
| 9 | LiHMDS | CH2Cl2 | 1.3 | 0 | 2.5 | 52 | 68:32 |
| 10 | LiHMDS | CH2Cl2 | 1.3 | rt | 2.5 | 38 | 63:37 |
| 11 | LiHMDS | CH2Cl2 | 1.3 | −78 | 1.0 | 62 | 67:33 |
| 12 | LiHMDS | CH2Cl2 | 1.3 | −78 | 1.5 | 82 | 67:33 |
| 13d | LiHMDS | CH2Cl2 | 1.3 | −78 | 2.5 | 65 | 66:34 |
| 14e | LiHMDS | CH2Cl2 | 1.3 | −78 | 2.5 | 66 | 70:30 |
aReaction conditions: 1 (0.3 mmol), base (1.3 equiv of 2a), solvent (3 mL), under nitrogen. bIsolated yield of two isomers. cDetermined by 19F NMR. d0.5 equiv of BF3·Et2O was added. e0.5 equiv of Ti(OiPr)4 was added.
With the optimized reaction conditions in hand, we examined the generality of these asymmetric Mannich reactions by using various arylethynes 2 (Scheme 2). Under standard reaction conditions, all the tested substrates worked well to generate the corresponding Mannich adducts in moderate to good yields. In particular, ethynylbenzenes bearing electron-donating substituents on the aromatic ring were more compatible with this reaction, resulting in higher chemical yields (53–87%) and better diastereoselectivities (3a–g). The ethynylbenzene 2g featuring a long alkyl chain on para-position was also a suitable substrate, which was converted into product 3g in 77% yield. On the other hand, arylethynes bearing electron-withdrawing groups on the aromatic ring showed lower reactivity along with lower yields (31–55%) as well as poorer diastereoselectivities (3i–m), except for 3n which gave 70:30 dr. It should be mentioned that the position of substituents showed almost no effects on the reaction efficiency (3c vs 3d, 3i vs 3j, 3l vs 3m). Also, importantly, two diastereomers, in all the cases 3a–n, can be readily obtained in diastereomerically pure form by routine column chromatography, underscoring the value of this strategy for the synthesis of chiral α-trifluoromethylated propargylamines.
![[1860-5397-16-217-i2]](/bjoc/content/inline/1860-5397-16-217-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope study. Reaction conditions: arylethyne 2 (0.39 mmol), imine 1 (0.3 mmol), LiHMDS (0.51 mmol), CH2Cl2 (3 mL), −78 °C, under nitrogen, 2.5 h. Isolated yields of mixture of isomers. Diastereoselectivities were determined by 19F NMR.
Scheme 2: Substrate scope study. Reaction conditions: arylethyne 2 (0.39 mmol), imine 1 (0.3 mmol), LiHMDS (0...
In order to determine the absolute configuration of the chiral addition products, we successfully performed a crystallographic analysis of the minor product 3a. The structure is shown in Figure 2, the absolute configuration of the newly generated chiral center of the minor product 3a is S, which demonstrates accordingly that the major one is R. The absolute configurations of other corresponding products were assigned by analogy.
![[1860-5397-16-217-2]](/bjoc/content/figures/1860-5397-16-217-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram showing of the minor product of 3a.
Figure 2: ORTEP diagram showing of the minor product of 3a.
In general, the low-to-moderate diastereoselectivity observed in this study was unexpected. In fact, considering the excellent level (>90% de) of stereocontrol reported for the Mannich additions of aldimine 1 with sp3 [42,55] and sp2 [56] nucleophiles, including lithiated aromatics [57] one would not anticipate such a dramatic drop in the selectivity in the reactions of sp nucleophiles. Accordingly, we considered the mode of nucleophilic attack on the imine double bond in 1. As presented in Figure 3, imine 1 might react in the most stable conformation with the bulky tert-butyl group pointing away from the imine double bond. In this case the only stereocontrolling factor in play is the difference between the sulfinyl oxygen and the oxygen electron lone pair. The latter presents a lesser steric obstacle rendering the corresponding nucleophilic attack the more probable event.
![[1860-5397-16-217-3]](/bjoc/content/figures/1860-5397-16-217-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Mode of nucleophilic attacks A and B.
Figure 3: Mode of nucleophilic attacks A and B.
These ever-unexpected results strongly suggest that the origin of the stereocontrol in the reactions of imine 1 is not just the bulk of the tert-butyl group but, as a major factor, the corresponding chelated transition states.
As all diastereomers 3 can be easily isolated in optical pure form by routine column chromatography, this reaction should be of certain value for biological studies. In this regard, we decided to demonstrate the reproducibility of this method for large-scale synthesis (Scheme 3) and the removal of the sulfinyl auxiliary to give the free amine (Scheme 4). To our delight, 1.4 g of the desired product 3a was obtained when the amount of sulfinylimine 1 was raised to 1.0 g (5.0 mmol). Comparing with reaction of 0.3 mmol scale, only a slight decrease in the yield was observed (84%) along with the same stereochemical outcome. Also, the chiral sulfinyl auxiliary can be easily removed under acidic conditions to give the free amine. Treating 3i with HCl gas in methanol at 0 °C, the tert-butylsulfinyl group was cleaved under mild conditions and the target primary amine 4 was obtained with 57% yield.
![[1860-5397-16-217-i3]](/bjoc/content/inline/1860-5397-16-217-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Large-scale application of the reaction.
Scheme 3: Large-scale application of the reaction.
![[1860-5397-16-217-i4]](/bjoc/content/inline/1860-5397-16-217-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Removal of the chiral auxiliary.
Scheme 4: Removal of the chiral auxiliary.
Conclusion
In conclusion, we have explored arylethynes as new nucleophiles for the Mannich reactions of (R)-N-tert-butylsulfinyl-3,3,3-trifluoroacetaldimine. Quite unexpectedly, the diastereoselectivity of the reactions were noticeably lower when compared with the corresponding reactions of sp3 and sp2 nucleophiles. Nevertheless, the two diastereomers can be easily isolated in diastereomerically pure state by regular column chromatography. This method provides access to chiral trifluoromethylpropargylamines. The application of these compounds for the synthesis of biologically relevant targets and their self-disproportionation of enantiomers (SDE) properties [58-60] are currently under study and will be reported in due course.
Experimental
General procedure for the Mannich reaction of arylethynes with sulfinylimine: Into an oven-dried reaction vial flushed with N2 were taken the respective arylethyne 2 (0.39 mmol) and anhydrous CH2Cl2 (2.0 mL). The reaction vial was cooled to −78 °C and LiHMDS (1 M in THF, 0.51 mmol) was added dropwise with stirring. After 1 h at −78 °C, sulfinylimine 1 (0.3 mmol), dissolved in anhydrous CH2Cl2 (1.0 mL), was added dropwise. Stirring was continued at −78 °C for 2.5 h, then the reaction was quenched with saturated NH4Cl (2.0 mL), followed by H2O (5.0 mL) and the mixture was brought to room temperature. The organic layer was taken and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered and the solvent was removed to give the crude product 3, which was purified by column chromatography using hexane/EtOAc (4:1, v/v) as eluent.
Supporting Information
| Supporting Information File 1: Experimental details and spectral data. | ||
| Format: PDF | Size: 5.9 MB | Download |
References
-
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006
Return to citation in text: [1] -
O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003
Return to citation in text: [1] -
Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050
Return to citation in text: [1] -
Mei, H.; Han, J.; Klika, K. D.; Izawa, K.; Sato, T.; Meanwell, N. A.; Soloshonok, V. A. Eur. J. Med. Chem. 2020, 186, 111826. doi:10.1016/j.ejmech.2019.111826
Return to citation in text: [1] -
Mei, H.; Han, J.; White, S.; Graham, D. J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N. A.; Soloshonok, V. A. Chem. – Eur. J. 2020, 26, 11349–11390. doi:10.1002/chem.202000617
Return to citation in text: [1] -
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
Return to citation in text: [1] -
Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014
Return to citation in text: [1] -
Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011
Return to citation in text: [1] -
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
Return to citation in text: [1] -
Aceña, J. L.; Sorochinsky, A. E.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 155, 21–38. doi:10.1016/j.jfluchem.2013.06.004
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Zhang, W. Chem. Rev. 2009, 109, 749–795. doi:10.1021/cr800412s
Return to citation in text: [1] -
Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f
Return to citation in text: [1] -
Hagiwara, R.; Ito, Y. J. Fluorine Chem. 2000, 105, 221–227. doi:10.1016/s0022-1139(99)00267-5
Return to citation in text: [1] -
Fang, M.; Okamoto, Y.; Koike, Y.; He, Z.; Merkel, T. C. J. Fluorine Chem. 2016, 188, 18–22. doi:10.1016/j.jfluchem.2016.05.013
Return to citation in text: [1] -
Amatucci, G. G.; Pereira, N. J. Fluorine Chem. 2007, 128, 243–262. doi:10.1016/j.jfluchem.2006.11.016
Return to citation in text: [1] -
Hoepping, A.; Johnson, K. M.; George, C.; Flippen-Anderson, J.; Kozikowski, A. P. J. Med. Chem. 2000, 43, 2064–2071. doi:10.1021/jm0001121
Return to citation in text: [1] -
Davidson, M. H.; McDonald, F. E. Org. Lett. 2004, 6, 1601–1603. doi:10.1021/ol049630m
Return to citation in text: [1] -
Trost, B. M.; Chung, C. K.; Pinkerton, A. B. Angew. Chem., Int. Ed. 2004, 43, 4327–4329. doi:10.1002/anie.200460058
Return to citation in text: [1] -
Fleming, J. J.; Du Bois, J. J. Am. Chem. Soc. 2006, 128, 3926–3927. doi:10.1021/ja0608545
Return to citation in text: [1] -
Llobat, A.; Escorihuela, J.; Sedgwick, D. M.; Rodenes, M.; Román, R.; Soloshonok, V. A.; Han, J.; Medio-Simón, M.; Fustero, S. Eur. J. Org. Chem. 2020, 4193–4207. doi:10.1002/ejoc.202000598
Return to citation in text: [1] -
Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J. L.; Izawa, K.; Liu, H.; Soloshonok, V. A. J. Fluorine Chem. 2014, 167, 37–54. doi:10.1016/j.jfluchem.2014.06.026
Return to citation in text: [1] -
Huang, Y.-Y.; Yang, X.; Chen, Z.; Verpoort, F.; Shibata, N. Chem. – Eur. J. 2015, 21, 8664–8684. doi:10.1002/chem.201500361
Return to citation in text: [1] -
Kauffman, G. S.; Harris, G. D.; Dorow, R. L.; Stone, B. R. P.; Parsons, R. L.; Pesti, J. A.; Magnus, N. A.; Fortunak, J. M.; Confalone, P. N.; Nugent, W. A. Org. Lett. 2000, 2, 3119–3121. doi:10.1021/ol006321x
Return to citation in text: [1] -
Crucianelli, M.; Angelis, F. D.; Lazzaro, F.; Malpezzi, L.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 573–577. doi:10.1016/j.jfluchem.2003.11.034
Return to citation in text: [1] -
Jiang, B.; Si, Y.-G. Angew. Chem., Int. Ed. 2004, 43, 216–218. doi:10.1002/anie.200352301
Return to citation in text: [1] -
Zhang, F.-G.; Ma, H.; Nie, J.; Zheng, Y.; Gao, Q.; Ma, J.-A. Adv. Synth. Catal. 2012, 354, 1422–1428. doi:10.1002/adsc.201100926
Return to citation in text: [1] -
Zhang, F.-G.; Ma, H.; Zheng, Y.; Ma, J.-A. Tetrahedron 2012, 68, 7663–7669. doi:10.1016/j.tet.2012.05.086
Return to citation in text: [1] -
Morisaki, K.; Sawa, M.; Nomaguchi, J.-y.; Morimoto, H.; Takeuchi, Y.; Mashima, K.; Ohshima, T. Chem. – Eur. J. 2013, 19, 8417–8420. doi:10.1002/chem.201301237
Return to citation in text: [1] -
Chen, M.-W.; Wu, B.; Chen, Z.-P.; Shi, L.; Zhou, Y.-G. Org. Lett. 2016, 18, 4650–4653. doi:10.1021/acs.orglett.6b02283
Return to citation in text: [1] -
Chen, M.-W.; Yang, Q.; Deng, Z.; Zhou, Y.; Ding, Q.; Peng, Y. J. Org. Chem. 2018, 83, 8688–8694. doi:10.1021/acs.joc.8b00873
Return to citation in text: [1] -
Xiao, H.; Huang, Y.; Qing, F.-L. Tetrahedron: Asymmetry 2010, 21, 2949–2955. doi:10.1016/j.tetasy.2010.11.028
Return to citation in text: [1] -
Xie, C.; Wu, L.; Han, J.; Soloshonok, V. A.; Pan, Y. Angew. Chem., Int. Ed. 2015, 54, 6019–6023. doi:10.1002/anie.201500908
Return to citation in text: [1] -
Han, J.; Sorochinsky, A. E.; Ono, T.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 281–294. doi:10.2174/157017911794697277
Return to citation in text: [1] -
Xie, C.; Zhang, L.; Sha, W.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Lett. 2016, 18, 3270–3273. doi:10.1021/acs.orglett.6b01516
Return to citation in text: [1] -
Xie, C.; Dai, Y.; Mei, H.; Han, J.; Soloshonok, V. A.; Pan, Y. Chem. Commun. 2015, 51, 9149–9152. doi:10.1039/c5cc02256h
Return to citation in text: [1] -
Xie, C.; Wu, L.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. Tetrahedron Lett. 2014, 55, 5908–5910. doi:10.1016/j.tetlet.2014.09.001
Return to citation in text: [1] -
Xie, C.; Wu, L.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 7836–7843. doi:10.1039/c4ob01575d
Return to citation in text: [1] [2] -
Mei, H.; Xiong, Y.; Han, J.; Pan, Y. Org. Biomol. Chem. 2011, 9, 1402–1406. doi:10.1039/c0ob00586j
Return to citation in text: [1] [2] [3] -
Xie, C.; Mei, H.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. RSC Adv. 2014, 4, 4763–4768. doi:10.1039/c3ra45773g
Return to citation in text: [1] [2] -
Mei, H.; Xie, C.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2013, 11, 8018–8021. doi:10.1039/c3ob41785a
Return to citation in text: [1] [2] -
Mei, H.; Xiong, Y.; Xie, C.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 2108–2113. doi:10.1039/c3ob42348d
Return to citation in text: [1] [2] -
Liu, Y.; Huang, Y.; Qing, F.-L. Tetrahedron 2012, 68, 4955–4961. doi:10.1016/j.tet.2012.04.070
Return to citation in text: [1] [2] -
Liu, Y.; Yang, Y.; Huang, Y.; Xu, X.-H.; Qing, F. L. Synlett 2015, 26, 67–72. doi:10.1055/s-0034-1379600
Return to citation in text: [1] [2] -
Shibata, N.; Nishimine, T.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Sorochinsky, A. E.; Soloshonok, V. A. Chem. Commun. 2012, 48, 4124–4126. doi:10.1039/c2cc30627a
Return to citation in text: [1] [2] -
Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578
Return to citation in text: [1] [2] -
Mei, H.; Han, J.; Fustero, S.; Román, R.; Ruzziconi, R.; Soloshonok, V. A. J. Fluorine Chem. 2018, 216, 57–70. doi:10.1016/j.jfluchem.2018.10.003
Return to citation in text: [1] [2] -
Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t
Return to citation in text: [1] [2] -
Liu, P.; Liu, Z.-J.; Wu, F. Adv. Synth. Catal. 2015, 357, 818–822. doi:10.1002/adsc.201400992
Return to citation in text: [1] [2] -
Boerth, J. A.; Hummel, J. R.; Ellman, J. A. Angew. Chem., Int. Ed. 2016, 55, 12650–12654. doi:10.1002/anie.201603831
Return to citation in text: [1] [2] -
Sanz-Vidal, Á.; Torres, J.; Soloshonok, V. A.; Zhu, Y.; Han, J.; Fustero, S.; del Pozo, C. Adv. Synth. Catal. 2018, 360, 366–373. doi:10.1002/adsc.201701284
Return to citation in text: [1] [2] -
Shevchuk, M. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Bassil, B. S.; Kawada, K.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6479. doi:10.1039/c3ra40687c
Return to citation in text: [1] -
Wu, L.; Xie, C.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. J. Org. Chem. 2014, 79, 7677–7681. doi:10.1021/jo5012009
Return to citation in text: [1] -
Mei, H.; Dai, Y.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Eur. J. Org. Chem. 2014, 2429–2433. doi:10.1002/ejoc.201400118
Return to citation in text: [1] -
Sorochinsky, A. E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J. L.; Soloshonok, V. A. Chirality 2013, 25, 365–368. doi:10.1002/chir.22180
Return to citation in text: [1] -
Sorochinsky, A. E.; Aceña, J. L.; Soloshonok, V. A. Synthesis 2013, 45, 141–152. doi:10.1055/s-0032-1316812
Return to citation in text: [1] -
Han, J.; Nelson, D. J.; Sorochinsky, A. E.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 310–317. doi:10.2174/157017911794697303
Return to citation in text: [1]
| 58. | Sorochinsky, A. E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J. L.; Soloshonok, V. A. Chirality 2013, 25, 365–368. doi:10.1002/chir.22180 |
| 59. | Sorochinsky, A. E.; Aceña, J. L.; Soloshonok, V. A. Synthesis 2013, 45, 141–152. doi:10.1055/s-0032-1316812 |
| 60. | Han, J.; Nelson, D. J.; Sorochinsky, A. E.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 310–317. doi:10.2174/157017911794697303 |
| 1. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 2. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 3. | Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007 |
| 4. | Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006 |
| 5. | O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003 |
| 6. | Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050 |
| 7. | Mei, H.; Han, J.; Klika, K. D.; Izawa, K.; Sato, T.; Meanwell, N. A.; Soloshonok, V. A. Eur. J. Med. Chem. 2020, 186, 111826. doi:10.1016/j.ejmech.2019.111826 |
| 8. | Mei, H.; Han, J.; White, S.; Graham, D. J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N. A.; Soloshonok, V. A. Chem. – Eur. J. 2020, 26, 11349–11390. doi:10.1002/chem.202000617 |
| 9. | Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778 |
| 25. | Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J. L.; Izawa, K.; Liu, H.; Soloshonok, V. A. J. Fluorine Chem. 2014, 167, 37–54. doi:10.1016/j.jfluchem.2014.06.026 |
| 26. | Huang, Y.-Y.; Yang, X.; Chen, Z.; Verpoort, F.; Shibata, N. Chem. – Eur. J. 2015, 21, 8664–8684. doi:10.1002/chem.201500361 |
| 56. | Wu, L.; Xie, C.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. J. Org. Chem. 2014, 79, 7677–7681. doi:10.1021/jo5012009 |
| 20. | Hoepping, A.; Johnson, K. M.; George, C.; Flippen-Anderson, J.; Kozikowski, A. P. J. Med. Chem. 2000, 43, 2064–2071. doi:10.1021/jm0001121 |
| 21. | Davidson, M. H.; McDonald, F. E. Org. Lett. 2004, 6, 1601–1603. doi:10.1021/ol049630m |
| 22. | Trost, B. M.; Chung, C. K.; Pinkerton, A. B. Angew. Chem., Int. Ed. 2004, 43, 4327–4329. doi:10.1002/anie.200460058 |
| 23. | Fleming, J. J.; Du Bois, J. J. Am. Chem. Soc. 2006, 128, 3926–3927. doi:10.1021/ja0608545 |
| 24. | Llobat, A.; Escorihuela, J.; Sedgwick, D. M.; Rodenes, M.; Román, R.; Soloshonok, V. A.; Han, J.; Medio-Simón, M.; Fustero, S. Eur. J. Org. Chem. 2020, 4193–4207. doi:10.1002/ejoc.202000598 |
| 57. | Mei, H.; Dai, Y.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Eur. J. Org. Chem. 2014, 2429–2433. doi:10.1002/ejoc.201400118 |
| 15. | Zhang, W. Chem. Rev. 2009, 109, 749–795. doi:10.1021/cr800412s |
| 16. | Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f |
| 17. | Hagiwara, R.; Ito, Y. J. Fluorine Chem. 2000, 105, 221–227. doi:10.1016/s0022-1139(99)00267-5 |
| 18. | Fang, M.; Okamoto, Y.; Koike, Y.; He, Z.; Merkel, T. C. J. Fluorine Chem. 2016, 188, 18–22. doi:10.1016/j.jfluchem.2016.05.013 |
| 19. | Amatucci, G. G.; Pereira, N. J. Fluorine Chem. 2007, 128, 243–262. doi:10.1016/j.jfluchem.2006.11.016 |
| 41. | Xie, C.; Wu, L.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 7836–7843. doi:10.1039/c4ob01575d |
| 42. | Mei, H.; Xiong, Y.; Han, J.; Pan, Y. Org. Biomol. Chem. 2011, 9, 1402–1406. doi:10.1039/c0ob00586j |
| 43. | Xie, C.; Mei, H.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. RSC Adv. 2014, 4, 4763–4768. doi:10.1039/c3ra45773g |
| 44. | Mei, H.; Xie, C.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2013, 11, 8018–8021. doi:10.1039/c3ob41785a |
| 45. | Mei, H.; Xiong, Y.; Xie, C.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 2108–2113. doi:10.1039/c3ob42348d |
| 46. | Liu, Y.; Huang, Y.; Qing, F.-L. Tetrahedron 2012, 68, 4955–4961. doi:10.1016/j.tet.2012.04.070 |
| 47. | Liu, Y.; Yang, Y.; Huang, Y.; Xu, X.-H.; Qing, F. L. Synlett 2015, 26, 67–72. doi:10.1055/s-0034-1379600 |
| 48. | Shibata, N.; Nishimine, T.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Sorochinsky, A. E.; Soloshonok, V. A. Chem. Commun. 2012, 48, 4124–4126. doi:10.1039/c2cc30627a |
| 49. | Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578 |
| 50. | Mei, H.; Han, J.; Fustero, S.; Román, R.; Ruzziconi, R.; Soloshonok, V. A. J. Fluorine Chem. 2018, 216, 57–70. doi:10.1016/j.jfluchem.2018.10.003 |
| 51. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 52. | Liu, P.; Liu, Z.-J.; Wu, F. Adv. Synth. Catal. 2015, 357, 818–822. doi:10.1002/adsc.201400992 |
| 53. | Boerth, J. A.; Hummel, J. R.; Ellman, J. A. Angew. Chem., Int. Ed. 2016, 55, 12650–12654. doi:10.1002/anie.201603831 |
| 54. | Sanz-Vidal, Á.; Torres, J.; Soloshonok, V. A.; Zhu, Y.; Han, J.; Fustero, S.; del Pozo, C. Adv. Synth. Catal. 2018, 360, 366–373. doi:10.1002/adsc.201701284 |
| 10. | Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014 |
| 11. | Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011 |
| 12. | Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f |
| 13. | Aceña, J. L.; Sorochinsky, A. E.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 155, 21–38. doi:10.1016/j.jfluchem.2013.06.004 |
| 14. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 42. | Mei, H.; Xiong, Y.; Han, J.; Pan, Y. Org. Biomol. Chem. 2011, 9, 1402–1406. doi:10.1039/c0ob00586j |
| 55. | Shevchuk, M. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Bassil, B. S.; Kawada, K.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6479. doi:10.1039/c3ra40687c |
| 34. | Chen, M.-W.; Yang, Q.; Deng, Z.; Zhou, Y.; Ding, Q.; Peng, Y. J. Org. Chem. 2018, 83, 8688–8694. doi:10.1021/acs.joc.8b00873 |
| 36. | Xie, C.; Wu, L.; Han, J.; Soloshonok, V. A.; Pan, Y. Angew. Chem., Int. Ed. 2015, 54, 6019–6023. doi:10.1002/anie.201500908 |
| 37. | Han, J.; Sorochinsky, A. E.; Ono, T.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 281–294. doi:10.2174/157017911794697277 |
| 38. | Xie, C.; Zhang, L.; Sha, W.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Lett. 2016, 18, 3270–3273. doi:10.1021/acs.orglett.6b01516 |
| 39. | Xie, C.; Dai, Y.; Mei, H.; Han, J.; Soloshonok, V. A.; Pan, Y. Chem. Commun. 2015, 51, 9149–9152. doi:10.1039/c5cc02256h |
| 40. | Xie, C.; Wu, L.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. Tetrahedron Lett. 2014, 55, 5908–5910. doi:10.1016/j.tetlet.2014.09.001 |
| 33. | Chen, M.-W.; Wu, B.; Chen, Z.-P.; Shi, L.; Zhou, Y.-G. Org. Lett. 2016, 18, 4650–4653. doi:10.1021/acs.orglett.6b02283 |
| 41. | Xie, C.; Wu, L.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 7836–7843. doi:10.1039/c4ob01575d |
| 42. | Mei, H.; Xiong, Y.; Han, J.; Pan, Y. Org. Biomol. Chem. 2011, 9, 1402–1406. doi:10.1039/c0ob00586j |
| 43. | Xie, C.; Mei, H.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. RSC Adv. 2014, 4, 4763–4768. doi:10.1039/c3ra45773g |
| 44. | Mei, H.; Xie, C.; Wu, L.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2013, 11, 8018–8021. doi:10.1039/c3ob41785a |
| 45. | Mei, H.; Xiong, Y.; Xie, C.; Soloshonok, V. A.; Han, J.; Pan, Y. Org. Biomol. Chem. 2014, 12, 2108–2113. doi:10.1039/c3ob42348d |
| 46. | Liu, Y.; Huang, Y.; Qing, F.-L. Tetrahedron 2012, 68, 4955–4961. doi:10.1016/j.tet.2012.04.070 |
| 47. | Liu, Y.; Yang, Y.; Huang, Y.; Xu, X.-H.; Qing, F. L. Synlett 2015, 26, 67–72. doi:10.1055/s-0034-1379600 |
| 48. | Shibata, N.; Nishimine, T.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Sorochinsky, A. E.; Soloshonok, V. A. Chem. Commun. 2012, 48, 4124–4126. doi:10.1039/c2cc30627a |
| 49. | Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578 |
| 50. | Mei, H.; Han, J.; Fustero, S.; Román, R.; Ruzziconi, R.; Soloshonok, V. A. J. Fluorine Chem. 2018, 216, 57–70. doi:10.1016/j.jfluchem.2018.10.003 |
| 51. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 52. | Liu, P.; Liu, Z.-J.; Wu, F. Adv. Synth. Catal. 2015, 357, 818–822. doi:10.1002/adsc.201400992 |
| 53. | Boerth, J. A.; Hummel, J. R.; Ellman, J. A. Angew. Chem., Int. Ed. 2016, 55, 12650–12654. doi:10.1002/anie.201603831 |
| 54. | Sanz-Vidal, Á.; Torres, J.; Soloshonok, V. A.; Zhu, Y.; Han, J.; Fustero, S.; del Pozo, C. Adv. Synth. Catal. 2018, 360, 366–373. doi:10.1002/adsc.201701284 |
| 28. | Crucianelli, M.; Angelis, F. D.; Lazzaro, F.; Malpezzi, L.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 573–577. doi:10.1016/j.jfluchem.2003.11.034 |
| 29. | Jiang, B.; Si, Y.-G. Angew. Chem., Int. Ed. 2004, 43, 216–218. doi:10.1002/anie.200352301 |
| 30. | Zhang, F.-G.; Ma, H.; Nie, J.; Zheng, Y.; Gao, Q.; Ma, J.-A. Adv. Synth. Catal. 2012, 354, 1422–1428. doi:10.1002/adsc.201100926 |
| 31. | Zhang, F.-G.; Ma, H.; Zheng, Y.; Ma, J.-A. Tetrahedron 2012, 68, 7663–7669. doi:10.1016/j.tet.2012.05.086 |
| 32. | Morisaki, K.; Sawa, M.; Nomaguchi, J.-y.; Morimoto, H.; Takeuchi, Y.; Mashima, K.; Ohshima, T. Chem. – Eur. J. 2013, 19, 8417–8420. doi:10.1002/chem.201301237 |
| 27. | Kauffman, G. S.; Harris, G. D.; Dorow, R. L.; Stone, B. R. P.; Parsons, R. L.; Pesti, J. A.; Magnus, N. A.; Fortunak, J. M.; Confalone, P. N.; Nugent, W. A. Org. Lett. 2000, 2, 3119–3121. doi:10.1021/ol006321x |
| 35. | Xiao, H.; Huang, Y.; Qing, F.-L. Tetrahedron: Asymmetry 2010, 21, 2949–2955. doi:10.1016/j.tetasy.2010.11.028 |
© 2020 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)