Abstract
The tandem gold(I)-catalyzed rearrangement/Nazarov reaction of enynyl acetates in which the double bond is embedded in a piperidine ring was computationally and experimentally studied. The theoretical calculations predict that the position of the propargylic acetate substituent has a great impact on the reactivity. In contrast to our previous successful cyclization of the 2-substituted substrates, where the nitrogen favors the formation of the cyclized final product, the substitution at position 3 was computed to have a deleterious effect on the electronic properties of the molecules, increasing the activation barriers of the Nazarov reaction. The sluggish reactivity of 3-substituted piperidines predicted by the calculations was further confirmed by the results obtained with some designed substrates.

Graphical Abstract
Introduction
In the development of new and effective catalysts, step economy is surely one of the major goals. A reduction of the number of steps in the synthesis of complex compounds can be attained by cascade reactions, which allow for structural modifications on the organic compounds by forming several chemical bonds in one pot. To this end, gold catalysis [1-7] has been widely exploited to construct various cyclic and heterocyclic frameworks through cascade reactions triggered by the activation of a triple bond, which has ultimately led to the total synthesis of several natural compounds [2,8]. The gold-catalyzed rearrangement of suitably substituted propargylic esters in particular provides a platform for cascade processes that involve a cationic or an allene intermediate generated in the first step [1,9-12].
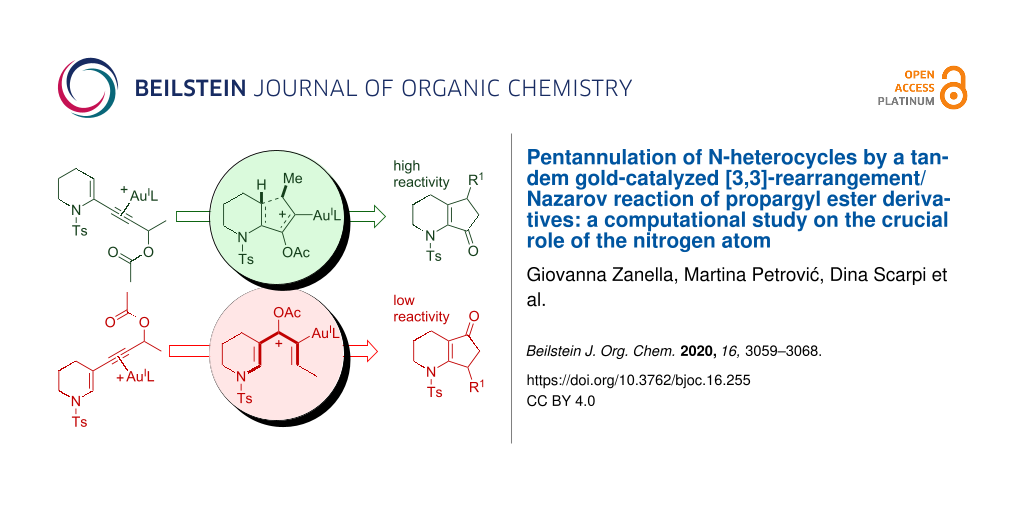
In the framework of our studies on gold(I)-catalyzed reactions of propargyl alcohol derivatives [13-15], we have recently reported that the pentannulation of N-heterocycles [16] can be efficiently achieved by a cascade gold-catalyzed [3,3]-rearrangement/Nazarov reaction of propargyl ester derivatives (Figure 1a) [17-24], and we have exploited such a methodology for the synthesis of bruceollines H and I from 3-substituted indoles (Figure 1b) [25,26]. Our computational study showed that the Nazarov reaction is fast with the 2-substituted piperidine derivatives 1 because of the accelerating effect of the nitrogen atom that stabilizes the oxyallyl cation intermediate 4 formed upon the ring closure. This was in analogy to that found for the classical Brønsted or Lewis acid-catalyzed Nazarov reaction involving N-heterocycles [27-37] and in accordance with the polarized Nazarov reaction concept developed by Frontier [27,33].
![[1860-5397-16-255-1]](/bjoc/content/figures/1860-5397-16-255-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tandem acetate rearrangement/Nazarov cyclization of different substrates.
Figure 1: Tandem acetate rearrangement/Nazarov cyclization of different substrates.
In an effort to broaden the scope of the reaction and the diversity of products, we assumed that the N-heterocycles 5, bearing the propargyl side chain at C3, would deliver a cyclopenta-fused heterocyclic system with an alternate position of the C=O group on the five-membered ring when treated with gold(I) (see 7, Figure 1c). In this context, the Nazarov cyclization has been profusely studied, and it was found that it is very sensitive to the electronic features of the substrates. For example, the rate is optimal in polarized systems obtained by the proper introduction of electronically asymmetric fragments [38,39]. Thus, an unsuitable combination of substituents can be detrimental for the reactivity, and we were aware that the electron donor nitrogen in 5 (having a side chain at C3) could stabilize the pentadienyl cationic intermediate 6, and thus relenting the 4π-electrocyclization, causing either the degradation of the starting material or the formation of unwanted side products. In fact, preliminary experimental results with 5 pointed in this direction, and we decided to carry out a complete computational analysis to evaluate the entire reaction profile and to help us validate our hypothesis before embarking on a potential total synthesis, involving such a process, in the future. In parallel, a few suitable substrates were also subjected to gold catalysis with the aim of verifying the conclusions drawn by the calculations.
Results and Discussion
Computational methods
In order to identify the structures and the energies of the critical steps of the mechanism, the potential reaction coordinates of the whole tandem [3,3]-rearrangement/Nazarov cyclization were studied computationally (Figure 2). To this end, a model substrate bearing p-toluensulfonyl as the protecting group on the nitrogen atom was chosen owing to the compatibility with such a process [16]. The structures were located using the B3LYP density functional theory method as implemented in the Gaussian suite of programs, using the 6-31G(d,p) basis set for nonmetallic atoms and SDD for Au. The alkynyl–gold(I) cationic complex I (Figure 2) was considered as the starting point of the mechanism (ΔG = 0 kcal⋅mol−1), and all reported energy values in the following discussion are relative to this figure. The values for ΔG correspond to the Gibbs energy computed at the M06/def2tzvpp level of theory in a solvent model (IEFPCM, solvent = DCM). The intrinsic reaction coordinates (IRC) were followed to verify the energy profiles connecting the key transition structures to the correct associated local minima. Ph3P was chosen as the ligand in analogy to the previous calculations on compound 1 [16].
![[1860-5397-16-255-2]](/bjoc/content/figures/1860-5397-16-255-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 3-substituted piperidine derivatives.
Figure 2: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 3...
Computational discussion
In analogy to similar processes [11,21,22,40,41], the reaction is initiated by a two-step [3,3]-acetate rearrangement [42], triggered by the coordination of the cationic gold species to the alkyne 5, as in I (Figure 2). The first step TS1 has a low activation energy (ΔG‡ = 12.2 kcal⋅mol−1) to form the unstable cyclic intermediate II. This short-lived species rapidly reopens through TS2 (ΔΔG‡ = 8.1 kcal⋅mol−1) to give the pentadienyl cation III, which presents a high stability (ΔG = −5.4 kcal⋅mol−1), and thus making the Nazarov cyclization through TS3 an endergonic process (from III to IV). In fact, the energy values calculated in Figure 2 show that either TS2 or TS3 or a combination of the two, depending on the reaction conditions, could be rate determining as they share very similar numbers, 14.6 kcal⋅mol−1 (from I to TS2) and 14.3 kcal⋅mol−1 (from III to TS3), respectively. We also computed the following steps of deprotonation, protodeauration, and acetate hydrolysis, which would lead to the final product 7, showing that they are not critical for the rate and outcome of the reaction. Thus, they will be discussed later separately.
Confirming our working hypothesis, this set of initial data contrasts with the computed gold(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 1. We had previously shown that for 2-substituted analogs of 1 (NCO2Me), the acetate rearrangement (specially the TS1-like first step). is rate determining with a low activation barrier of 10.0 kcal⋅mol−1 and that the Nazarov-cyclization is an extremely easy process (ΔΔG‡ = 5.1 kcal⋅mol−1) [16]. To homogenize with our results in Figure 1, we computed the corresponding N-sulfonyl-protected derivative 1 (Figure 3), confirming the differences that the 2- and 3-substitution, respectively, exert in the reaction outcome. Starting with V, the acetate rearrangement is rate determining (ΔΔG‡ = 14.2 kcal⋅mol−1), and more importantly, the activation energy for the cyclization in TS6 is very low (ΔΔG‡ = 7.2 kcal⋅mol−1) and highly exergonic (ΔΔG‡ = −17.5 kcal⋅mol−1), making the process from VII to VIII completely irreversible. In contrast, the 3-substituted intermediate III gives a much slower and reversible process.
![[1860-5397-16-255-3]](/bjoc/content/figures/1860-5397-16-255-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 2-substituted piperidine derivatives.
Figure 3: DFT-computed energy profile of the tandem Au(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 2...
Thus, the main reason for the worse performance of 5 as a substrate seems to be related to the higher stability of the intermediate III, which could be attributed to the π-donating ability of the nitrogen atom to stabilize the positive charge [43]. We evaluated this effect by calculating the charges through “natural bond orbital analysis” (NBO) of the atoms of the intermediate III and the 2-substitued analogue VII and found a significant difference between the two (Figure 4).
![[1860-5397-16-255-4]](/bjoc/content/figures/1860-5397-16-255-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Computed comparison of the NBO charges of 2- and 3-substituted substrates.
Figure 4: Computed comparison of the NBO charges of 2- and 3-substituted substrates.
Indeed, the total allyl charge on the 3-substituted intermediate III results to be almost neutral or even slightly negative (−0.056 e), confirming that the lone pair of the nitrogen atom can stabilize the positive charge of the allyl system by conjugation, affecting the following cyclization reaction. Meanwhile, the 2-substitued analogue VII does not present a conjugated system, and the total allyl charge cannot be stabilized, maintaining a positive value (+0.115). As a result, VII seems to be much more reactive and the associated cyclization much more exergonic than for III. The carbon atoms C1 and C3 seem to be especially more positive in VII than in III.
A second obvious difference between the two isomeric pathways in Figure 2 and Figure 3 is the much higher relative stability of the cyclized structure VIII compared to the analogue IV (−16.7 vs −0.9 kcal⋅mol−1). The π-donating ability of the nitrogen atom might have a clear stabilizing effect in VIII, while the nitrogen and the cationic allyl system are disconnected in IV. This effect is reflected in the corresponding transition states, with TS3 being higher in energy than TS6.
As mentioned before, after the slow cyclization step in TS3, we focused our analysis on the transformation of the bicyclic intermediate IV to the final diene product. Basically, the final steps have to include a deprotonation, protodeauration, and in some cases acetate hydrolysis. These steps can occur through different pathways; in particular, we considered a single-step intramolecular hydride shift with concomitant C–Au-bond breaking (Figure 5) or a base-mediated deprotonation, followed by Au–C-bond hydrolysis through protodeauration (Figure 6). In the former case, it emerged that the 1,2-hydrogen shift in TS7 is quite high in energy (ΔG‡ = 18.5 kcal⋅mol−1) relative to the previous barriers shown in Figure 2. This barrier is also much higher than the traditional 1,2-hydride shift in carbocations, which usually show barriers even under 10 kcal⋅mol−1. It has been suggested that the presence of water can catalyze this reaction (proton-transport catalysis strategy) through a two-step deprotonation/protonation process [11,21,41,42,44], but in our study, preliminary calculations in the presence of water did not improve the results in Figure 5.
![[1860-5397-16-255-5]](/bjoc/content/figures/1860-5397-16-255-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Single-step transformation of IV to IX.
Figure 5: Single-step transformation of IV to IX.
![[1860-5397-16-255-6]](/bjoc/content/figures/1860-5397-16-255-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Triflate-promoted hydrogen abstraction and protodeauration with HOTf.
Figure 6: Triflate-promoted hydrogen abstraction and protodeauration with HOTf.
Therefore, we focused on the proton abstraction pathway. Several possible bases exist in the reaction medium, such as the counterion in the gold(I) salt, the anion participating in the silver salt coadditives, or water. In analogy to our previous work [16], we initially modelled the deprotonation step with the triflate anion as a base (Figure 6).
In fact, the abstraction of the hydrogen atom in the position adjacent to Au (C3) shows a low activation barrier (TS8, ΔG‡ = 12.6 kcal⋅mol−1) from the corresponding precomplex, leading to the formation of the intermediate XI and triflic acid. The high acidity of the latter facilitates the protodeauration in the last step (TS9), which occurs exothermically and with a barrier of only 6.5 kcal⋅mol−1. The easiness of the two-step process from IV to XII is remarkable given the low basicity of the triflate anion, suggesting that other possible anions present in the medium could also play the same role. Comparing the different pathways in Figure 5 and Figure 6, it emerged that the base-mediated process is clearly favored over the 1,2-H-shift.
We were also aware of the regioselectivity issue that arose during the deprotonation due to the presence of two similar hydrogen atoms (Ha and Hb) in IV (Figure 7), and we wondered if there were significant differences between the two pathways. Indeed, the deprotonation of Ha does not seem as easy as Hb, despite the fact that the final product XV is a conjugated dienamine and more stable (ΔΔG = 4.4 kcal⋅mol−1) than XII, which lacks the conjugation. However, according to the energy profile, this observation does not have a reflection in the deprotonation step, which seems to be affected partially by the steric hindrance around the two hydrogen atoms, being clearly higher in Ha (a 2.2 kcal⋅mol−1 higher activation energy of TS10 than for TS8). Thus, under kinetic control, the reaction would lead to the formation of XII. However, as will be commented on later in the discussion, the experimental results clearly show the sole formation of compound 15 (Table 1), which arises from hydrolysis of an intermediate related to the complex XV. Thus, we believe that the higher thermodynamic stability of XV (4.4 kcal⋅mol−1 lower than for XII), which is due to the conjugation of the nitrogen atom and the diene system, accounts for the preferential formation and the consequent formation of 15. It cannot be overlooked that the formation of the intermediates XII and XV is hardly reversible due to the high exergonic character, and thus the equilibration of both final isomers through the previous intermediate IV is very unlikely. Our hypothesis is that an isomerization between XII and XV must be operative under these reaction conditions through a nonstudied protonation/deprotonation sequence. Finally, although we did not study in detail the acetate hydrolysis from XV to 15, we could confirm the higher stability (by more than 6 kcal⋅mol−1) of 15 relative to the enone isomer arising from XII, in agreement again with the experimental results.
![[1860-5397-16-255-7]](/bjoc/content/figures/1860-5397-16-255-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Triflate-mediated abstraction of the hydrogen atom Ha and protodeauration.
Figure 7: Triflate-mediated abstraction of the hydrogen atom Ha and protodeauration.
Table 1: Gold(I)-catalyzed [3,3]-rearrangement/Nazarov reaction of 14.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-255-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry | ligand | anion | time (h) | 15 (%)b | 16 (%) |
| 1 | PPh3 | TfO | 16 | 51 | 24 |
| 2 | PPh3 | SbF6 | 6 | 66 | c |
| 3 | PCy3 | SbF6 | 6 | 10d | – |
| 4 | P(4-CF3C6H4)3 | SbF6 | 6 | 49 | c |
| 5e | PPh3 | SbF6 | 1 | <5d | – |
| 6f | PPh3 | SbF6 | 4 | 48 | c |
aReaction conditions: 0.15–0.2 mmol of 14, 5 mol % of the catalyst, prepared by adding the silver salt to a 0.004 M solution of gold(I) chloride in DCM. The solvent was not dried before use unless otherwise indicated. bYield after chromatography unless otherwise indicated. cDetected by 1H NMR analysis of the crude reaction mixture. dConversion measured by 1H NMR. eUsing dry DCM. fReaction carried out in refluxing solvent.
Experimental discussion
As a summary of the previous discussion, we rationalized that the high stability of the intermediate III (especially compared to VII) and the relatively high activation energy of the cyclization in TS3 (vs the easier cyclization of TS6) could hamper the reactivity of 3-substituted piperidines, and that the slow cyclization of the intermediate III could result in starting material degradation or appearance of unwanted side reactions. To assess this hypothesis from an experimental point of view, the synthesis of the model compound 14 used in the calculation (as a gold complex I) was carried out and then subjected to gold catalysis. The synthesis started with the reduction of the N-Ts δ-valerolactam 8 with DIBAL-H into the corresponding lactamol 9 (Scheme 1), which was transformed into the enesulfonamide 10 in 70% yield by mesylation, followed by base-induced elimination of methanesulfonic acid, as previously reported [45]. In the next step, the electrophilic addition of iodine monochloride to the double bond of the enesulfonamide 10, followed by a nucleophilic attack of methanol on the formed iodonium ion afforded the α-methoxy-β-iodopiperidine 11 as a single stereoisomer (91% yield) [46]. The treatment of 11 with a catalytic amount of trifluoroacetic acid in toluene at 140 °C for 7 min led to the elimination of methanol and provided the 3-iodoenesulfonamide 12 in 77% yield [47]. To avoid the use of these harsh reaction conditions, we employed other methods, but both the iodination and bromination of 10 proved to be more difficult than anticipated. For example, attempts to obtain the 3-iodo derivative 11 using I2/Cs2CO3 in dioxane [48], NIS in DMF [49], NIS/AgNO3 in acetonitrile [50], and NIS/TFA in DCM [51] failed completely or provided the desired product as a complex mixture with unknown products. Then, the iodoenesulfonamide 12 was coupled with (±)-butyn-3-ol under Sonogashira conditions [52] to afford the enynyl alcohol 13, which was treated with acetic anhydride to provide the enynyl acetate 14 in a yield of 67% over two steps.
![[1860-5397-16-255-i1]](/bjoc/content/inline/1860-5397-16-255-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the enynyl acetate starting material 14.
Scheme 1: Synthesis of the enynyl acetate starting material 14.
Then, we applied the typical conditions for the [3,3]-rearrangement/Nazarov cyclization that we used for enynyl acetates of the type 1 (Figure 1) [16] to the enynyl acetate 14, i.e., 5 mol % Ph3PAuCl/AgOTf in DCM at room temperature (Table 1, entry 1) and 5 mol % of Ph3PAuCl/AgSbF6 in the same solvent (Table 1, entry 2), which were the best conditions that we tested in the rearrangement of the enynyl acetates 1 (e.g., with R = H, R1 = Me, EWG = Ts, the total yield of the Nazarov products was 85% after chromatography using AgSbF6 as the counter ion source, and with R = H, R1 = n-Bu, and EWG = Ts, the total yield was 86% when using AgOTf as the silver salt). Under both conditions, the reaction of 14 led to the formation of the cyclopentenone 15 in a lower yield (51% and 66%, respectively), and in comparison to the gold(I)-catalyzed reaction of the enynyl acetates 1, it was much slower with both catalytic systems (6–16 h vs 1.5–2 h for the complete disappearance of the starting material). Moreover, besides the ketone 16 [53], formed as byproduct in the reaction with AgOTf, we observed the formation of many other unidentified compounds, reasonably either via side reactions of gold intermediates or the degradation of the starting enynyl acetate 14.
In order to increase the reaction rate and decrease the amount of side products, the best reaction conditions (with AgSbF6) were modified by using different precatalysts (Table 1, entries 3 and 4), a dry solvent (Table 1, entry 5), and the reaction was also carried out at a higher temperature (Table 1, entry 6). However, none of these attempts were met with success, and indeed, a very sluggish reactivity was recorded in all these cases. Unfortunately, these results confirmed the negative predictions arising from the above calculations, but at the same time, they serve as a validation of the accuracy of the computational method we have used for the comparison of the isomeric complexes I and V.
Our previous experience in this area taught us that seven-membered azepane-derived enynyl acetates react faster than the corresponding piperidine analogues 1, prompting us to prepare enynyl acetate 20 as reported in Scheme 2. We wanted to confirm the negative effect of the substitution at the 3-position of the ring. We intended to follow a similar strategy to that outlined in Scheme 1, but the two-step procedure from 8 to 10 failed with the seven-membered ring. Thus, the enesulfonamide 18 was prepared via the palladium-catalyzed reduction of the corresponding phosphate 17 [54]. Iodination and Sonogashira coupling, followed by acetylation led to the formation of the desired enynyl acetate 20. This compound was treated with 5 mol % Ph3PAuCl/AgSbF6 in DCM, and after 6 h, this afforded the cyclopenta-fused product 21 in 54% yield. Again, the reaction was very slow compared to the corresponding 2-substituted azepane derivative and provided many unidentified side products, reducing our interest in the process.
![[1860-5397-16-255-i2]](/bjoc/content/inline/1860-5397-16-255-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and cyclization of enynyl acetate 20.
Scheme 2: Synthesis and cyclization of enynyl acetate 20.
Conclusion
In summary, we computationally studied and experimentally verified the [3,3]-rearrangement/Nazarov cyclization of 2,3-dehydropiperidines substituted with a propargyl acetate group in the 3-position, demonstrating the significance of the correct positioning of the nitrogen atom relative to the forming cycle. The comparison of the reactivity of the substrate having the piperidine ring substituted at the 2- vs the 3-position was very instructive about the optimal electronic features of the reactive species. In this regard, the initial rearrangement of the propargyl acetate induces the formation of a divinyl cationic intermediate, which is differently stabilized by conjugation with the nitrogen atom depending on the relative position of nitrogen. For the 3-substitution, the π-donor ability of the nitrogen atom strongly stabilizes the intermediate, reducing the reactivity. NBO calculations have also been used to confirm this hypothesis. We present some experimental data corroborating the sluggish reactivity of the 3-substituted substrates, in comparison to the 2-substituted analogues that we have previously described.
Supporting Information
| Supporting Information File 1: Computational section, experimental section, and NMR spectra. | ||
| Format: PDF | Size: 2.2 MB | Download |
References
-
Marín-Luna, M.; Nieto Faza, O.; Silva López, C. Front. Chem. (Lausanne, Switz.) 2019, 7, 296. doi:10.3389/fchem.2019.00296
Return to citation in text: [1] [2] -
Zi, W.; Toste, F. D. Chem. Soc. Rev. 2016, 45, 4567–4589. doi:10.1039/c5cs00929d
Return to citation in text: [1] [2] -
Michelet, V.; Toste, F. D., Eds. Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, U.K., 2014.
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Slaughter, L. M., Ed. Homogenous gold catalysis; Springer International Publishing: Basel, Switzerland, 2015.
Return to citation in text: [1] -
Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f
Return to citation in text: [1] -
Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773
Return to citation in text: [1] -
Marco-Contelles, J.; Soriano, E. Chem. – Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522
Return to citation in text: [1] [2] [3] -
Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769
Return to citation in text: [1] -
Rinaldi, A.; Langé, V.; Scarpi, D.; Occhiato, E. G. J. Org. Chem. 2020, 85, 5078–5086. doi:10.1021/acs.joc.0c00088
Return to citation in text: [1] -
Rinaldi, A.; Petrović, M.; Magnolfi, S.; Scarpi, D.; Occhiato, E. G. Org. Lett. 2018, 20, 4713–4717. doi:10.1021/acs.orglett.8b02141
Return to citation in text: [1] -
Rinaldi, A.; Langé, V.; Gómez-Bengoa, E.; Zanella, G.; Scarpi, D.; Occhiato, E. G. J. Org. Chem. 2019, 84, 6298–6311. doi:10.1021/acs.joc.9b00646
Return to citation in text: [1] -
Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lin, G.-Y.; Li, C.-W.; Hung, S.-H.; Liu, R.-S. Org. Lett. 2008, 10, 5059–5062. doi:10.1021/ol802047g
Return to citation in text: [1] -
Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417–16423. doi:10.1021/ja806415t
Return to citation in text: [1] -
Lemière, G.; Gandon, V.; Cariou, K.; Hours, A.; Fukuyama, T.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2009, 131, 2993–3006. doi:10.1021/ja808872u
Return to citation in text: [1] -
Lemière, G.; Gandon, V.; Cariou, K.; Fukuyama, T.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Org. Lett. 2007, 9, 2207–2209. doi:10.1021/ol070788r
Return to citation in text: [1] -
Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q
Return to citation in text: [1] [2] [3] -
Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503–15512. doi:10.1021/ja071070+
Return to citation in text: [1] [2] -
Congmon, J.; Tius, M. A. Eur. J. Org. Chem. 2018, 2926–2930. doi:10.1002/ejoc.201800604
Return to citation in text: [1] -
Hashmi, A. S. K.; Pankajakshan, S.; Rudolph, M.; Enns, E.; Bander, T.; Rominger, F.; Frey, W. Adv. Synth. Catal. 2009, 351, 2855–2875. doi:10.1002/adsc.200900614
Return to citation in text: [1] -
Scarpi, D.; Petrović, M.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Org. Lett. 2016, 18, 3922–3925. doi:10.1021/acs.orglett.6b01990
Return to citation in text: [1] -
Scarpi, D.; Faggi, C.; Occhiato, E. G. J. Nat. Prod. 2017, 80, 2384–2388. doi:10.1021/acs.jnatprod.7b00311
Return to citation in text: [1] -
Frontier, A. J.; Collison, C. Tetrahedron 2005, 61, 7577–7606. doi:10.1016/j.tet.2005.05.019
Return to citation in text: [1] [2] -
Tius, M. A. Eur. J. Org. Chem. 2005, 2193–2206. doi:10.1002/ejoc.200500005
Return to citation in text: [1] -
Wenz, D. R.; Read de Alaniz, J. Eur. J. Org. Chem. 2015, 23–37. doi:10.1002/ejoc.201402825
Return to citation in text: [1] -
Tius, M. A. Chem. Soc. Rev. 2014, 43, 2979–3002. doi:10.1039/c3cs60333d
Return to citation in text: [1] -
Di Grandi, M. J. Org. Biomol. Chem. 2014, 12, 5331–5345. doi:10.1039/c4ob00804a
Return to citation in text: [1] -
Spencer III, W. T.; Vaidya, T.; Frontier, A. J. Eur. J. Org. Chem. 2013, 3621–3633. doi:10.1002/ejoc.201300134
Return to citation in text: [1] -
Vaidya, T.; Eisenberg, R.; Frontier, A. J. ChemCatChem 2011, 3, 1531–1548. doi:10.1002/cctc.201100137
Return to citation in text: [1] [2] -
Occhiato, E. G.; Prandi, C.; Ferrali, A.; Guarna, A.; Venturello, P. J. Org. Chem. 2003, 68, 9728–9741. doi:10.1021/jo034939p
Return to citation in text: [1] -
Prandi, C.; Ferrali, A.; Guarna, A.; Venturello, P.; Occhiato, E. G. J. Org. Chem. 2004, 69, 7705–7709. doi:10.1021/jo0489263
Return to citation in text: [1] -
Larini, P.; Guarna, A.; Occhiato, E. G. Org. Lett. 2006, 8, 781–784. doi:10.1021/ol053071h
Return to citation in text: [1] -
Cavalli, A.; Pacetti, A.; Recanatini, M.; Prandi, C.; Scarpi, D.; Occhiato, E. G. Chem. – Eur. J. 2008, 14, 9292–9304. doi:10.1002/chem.200801030
Return to citation in text: [1] -
Frontier, A. J.; Hernandez, J. J. Acc. Chem. Res. 2020, 53, 1822–1832. doi:10.1021/acs.accounts.0c00284
Return to citation in text: [1] -
Riveira, M. J.; Marsili, L. A.; Mischne, M. P. Org. Biomol. Chem. 2017, 15, 9255–9274. doi:10.1039/c7ob02220d
Return to citation in text: [1] -
Miki, K.; Ohe, K.; Uemura, S. Tetrahedron Lett. 2003, 44, 2019–2022. doi:10.1016/s0040-4039(03)00219-3
Return to citation in text: [1] -
Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656–8657. doi:10.1021/ja0474695
Return to citation in text: [1] [2] -
The alternative acetate [3,2]-rearrangement was also computed, showing an activation energy of 19.6 kcal⋅mol−1. This pathway can be safely discarded since the corresponding energy is much higher than that of TS1 and TS2.
Return to citation in text: [1] [2] -
González-Pérez, A. B.; Villar, P.; de Lera, A. R. Eur. J. Org. Chem. 2019, 2539–2551. doi:10.1002/ejoc.201900103
Return to citation in text: [1] -
Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136
Return to citation in text: [1] -
Brown, D. S.; Charreau, P.; Hansson, T.; Ley, S. V. Tetrahedron 1991, 47, 1311–1328. doi:10.1016/s0040-4020(01)86388-2
Return to citation in text: [1] -
Norton Matos, M.; Afonso, C. A. M.; Batey, R. A. Tetrahedron 2005, 61, 1221–1244. doi:10.1016/j.tet.2004.11.035
Return to citation in text: [1] -
Zhang, H.; Hay, E. B.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2013, 135, 16610–16617. doi:10.1021/ja408387d
Return to citation in text: [1] -
Larivée, A.; Charette, A. B. Org. Lett. 2006, 8, 3955–3957. doi:10.1021/ol061415d
Return to citation in text: [1] -
Jana, S.; Rainier, J. D. Org. Lett. 2013, 15, 4426–4429. doi:10.1021/ol401974v
Return to citation in text: [1] -
Dharuman, S.; Vankar, Y. D. Org. Lett. 2014, 16, 1172–1175. doi:10.1021/ol500039s
Return to citation in text: [1] -
van den Broek, S. B. A. M. W.; Rensen, P. G. W.; van Delft, F. L.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 5906–5912. doi:10.1002/ejoc.201000587
Return to citation in text: [1] -
Kiewel, K.; Luo, Z.; Sulikowski, G. A. Org. Lett. 2005, 7, 5163–5165. doi:10.1021/ol051993e
Return to citation in text: [1] -
Compound 16 was easily identified by the α,β-unsaturated ketone moiety 1H NMR signals at 6.89 (dq, J = 15.2; 6.8 Hz), 6.63 (d, J = 15.2 Hz), and 1.94 (d, J = 6.8 Hz, CH3). The singlet at 5.45 ppm was diagnostic of 2-H in the 2,3-dehydropiperidines ring.
Return to citation in text: [1] -
Gigant, N.; Dequirez, G.; Retailleau, P.; Gillaizeau, I.; Dauban, P. Chem. – Eur. J. 2012, 18, 90–94. doi:10.1002/chem.201102302
Return to citation in text: [1]
| 53. | Compound 16 was easily identified by the α,β-unsaturated ketone moiety 1H NMR signals at 6.89 (dq, J = 15.2; 6.8 Hz), 6.63 (d, J = 15.2 Hz), and 1.94 (d, J = 6.8 Hz, CH3). The singlet at 5.45 ppm was diagnostic of 2-H in the 2,3-dehydropiperidines ring. |
| 54. | Gigant, N.; Dequirez, G.; Retailleau, P.; Gillaizeau, I.; Dauban, P. Chem. – Eur. J. 2012, 18, 90–94. doi:10.1002/chem.201102302 |
| 1. | Marín-Luna, M.; Nieto Faza, O.; Silva López, C. Front. Chem. (Lausanne, Switz.) 2019, 7, 296. doi:10.3389/fchem.2019.00296 |
| 2. | Zi, W.; Toste, F. D. Chem. Soc. Rev. 2016, 45, 4567–4589. doi:10.1039/c5cs00929d |
| 3. | Michelet, V.; Toste, F. D., Eds. Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, U.K., 2014. |
| 4. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 5. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 6. | Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454 |
| 7. | Slaughter, L. M., Ed. Homogenous gold catalysis; Springer International Publishing: Basel, Switzerland, 2015. |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 13. | Rinaldi, A.; Langé, V.; Scarpi, D.; Occhiato, E. G. J. Org. Chem. 2020, 85, 5078–5086. doi:10.1021/acs.joc.0c00088 |
| 14. | Rinaldi, A.; Petrović, M.; Magnolfi, S.; Scarpi, D.; Occhiato, E. G. Org. Lett. 2018, 20, 4713–4717. doi:10.1021/acs.orglett.8b02141 |
| 15. | Rinaldi, A.; Langé, V.; Gómez-Bengoa, E.; Zanella, G.; Scarpi, D.; Occhiato, E. G. J. Org. Chem. 2019, 84, 6298–6311. doi:10.1021/acs.joc.9b00646 |
| 43. | González-Pérez, A. B.; Villar, P.; de Lera, A. R. Eur. J. Org. Chem. 2019, 2539–2551. doi:10.1002/ejoc.201900103 |
| 1. | Marín-Luna, M.; Nieto Faza, O.; Silva López, C. Front. Chem. (Lausanne, Switz.) 2019, 7, 296. doi:10.3389/fchem.2019.00296 |
| 9. | Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d |
| 10. | Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773 |
| 11. | Marco-Contelles, J.; Soriano, E. Chem. – Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522 |
| 12. | Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769 |
| 11. | Marco-Contelles, J.; Soriano, E. Chem. – Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522 |
| 21. | Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q |
| 22. | Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503–15512. doi:10.1021/ja071070+ |
| 40. | Miki, K.; Ohe, K.; Uemura, S. Tetrahedron Lett. 2003, 44, 2019–2022. doi:10.1016/s0040-4039(03)00219-3 |
| 41. | Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656–8657. doi:10.1021/ja0474695 |
| 2. | Zi, W.; Toste, F. D. Chem. Soc. Rev. 2016, 45, 4567–4589. doi:10.1039/c5cs00929d |
| 8. | Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f |
| 42. | The alternative acetate [3,2]-rearrangement was also computed, showing an activation energy of 19.6 kcal⋅mol−1. This pathway can be safely discarded since the corresponding energy is much higher than that of TS1 and TS2. |
| 27. | Frontier, A. J.; Collison, C. Tetrahedron 2005, 61, 7577–7606. doi:10.1016/j.tet.2005.05.019 |
| 33. | Vaidya, T.; Eisenberg, R.; Frontier, A. J. ChemCatChem 2011, 3, 1531–1548. doi:10.1002/cctc.201100137 |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 27. | Frontier, A. J.; Collison, C. Tetrahedron 2005, 61, 7577–7606. doi:10.1016/j.tet.2005.05.019 |
| 28. | Tius, M. A. Eur. J. Org. Chem. 2005, 2193–2206. doi:10.1002/ejoc.200500005 |
| 29. | Wenz, D. R.; Read de Alaniz, J. Eur. J. Org. Chem. 2015, 23–37. doi:10.1002/ejoc.201402825 |
| 30. | Tius, M. A. Chem. Soc. Rev. 2014, 43, 2979–3002. doi:10.1039/c3cs60333d |
| 31. | Di Grandi, M. J. Org. Biomol. Chem. 2014, 12, 5331–5345. doi:10.1039/c4ob00804a |
| 32. | Spencer III, W. T.; Vaidya, T.; Frontier, A. J. Eur. J. Org. Chem. 2013, 3621–3633. doi:10.1002/ejoc.201300134 |
| 33. | Vaidya, T.; Eisenberg, R.; Frontier, A. J. ChemCatChem 2011, 3, 1531–1548. doi:10.1002/cctc.201100137 |
| 34. | Occhiato, E. G.; Prandi, C.; Ferrali, A.; Guarna, A.; Venturello, P. J. Org. Chem. 2003, 68, 9728–9741. doi:10.1021/jo034939p |
| 35. | Prandi, C.; Ferrali, A.; Guarna, A.; Venturello, P.; Occhiato, E. G. J. Org. Chem. 2004, 69, 7705–7709. doi:10.1021/jo0489263 |
| 36. | Larini, P.; Guarna, A.; Occhiato, E. G. Org. Lett. 2006, 8, 781–784. doi:10.1021/ol053071h |
| 37. | Cavalli, A.; Pacetti, A.; Recanatini, M.; Prandi, C.; Scarpi, D.; Occhiato, E. G. Chem. – Eur. J. 2008, 14, 9292–9304. doi:10.1002/chem.200801030 |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 25. | Scarpi, D.; Petrović, M.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Org. Lett. 2016, 18, 3922–3925. doi:10.1021/acs.orglett.6b01990 |
| 26. | Scarpi, D.; Faggi, C.; Occhiato, E. G. J. Nat. Prod. 2017, 80, 2384–2388. doi:10.1021/acs.jnatprod.7b00311 |
| 17. | Lin, G.-Y.; Li, C.-W.; Hung, S.-H.; Liu, R.-S. Org. Lett. 2008, 10, 5059–5062. doi:10.1021/ol802047g |
| 18. | Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417–16423. doi:10.1021/ja806415t |
| 19. | Lemière, G.; Gandon, V.; Cariou, K.; Hours, A.; Fukuyama, T.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2009, 131, 2993–3006. doi:10.1021/ja808872u |
| 20. | Lemière, G.; Gandon, V.; Cariou, K.; Fukuyama, T.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Org. Lett. 2007, 9, 2207–2209. doi:10.1021/ol070788r |
| 21. | Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q |
| 22. | Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503–15512. doi:10.1021/ja071070+ |
| 23. | Congmon, J.; Tius, M. A. Eur. J. Org. Chem. 2018, 2926–2930. doi:10.1002/ejoc.201800604 |
| 24. | Hashmi, A. S. K.; Pankajakshan, S.; Rudolph, M.; Enns, E.; Bander, T.; Rominger, F.; Frey, W. Adv. Synth. Catal. 2009, 351, 2855–2875. doi:10.1002/adsc.200900614 |
| 38. | Frontier, A. J.; Hernandez, J. J. Acc. Chem. Res. 2020, 53, 1822–1832. doi:10.1021/acs.accounts.0c00284 |
| 39. | Riveira, M. J.; Marsili, L. A.; Mischne, M. P. Org. Biomol. Chem. 2017, 15, 9255–9274. doi:10.1039/c7ob02220d |
| 45. | Brown, D. S.; Charreau, P.; Hansson, T.; Ley, S. V. Tetrahedron 1991, 47, 1311–1328. doi:10.1016/s0040-4020(01)86388-2 |
| 11. | Marco-Contelles, J.; Soriano, E. Chem. – Eur. J. 2007, 13, 1350–1357. doi:10.1002/chem.200601522 |
| 21. | Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q |
| 41. | Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656–8657. doi:10.1021/ja0474695 |
| 42. | The alternative acetate [3,2]-rearrangement was also computed, showing an activation energy of 19.6 kcal⋅mol−1. This pathway can be safely discarded since the corresponding energy is much higher than that of TS1 and TS2. |
| 44. | Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136 |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 52. | Kiewel, K.; Luo, Z.; Sulikowski, G. A. Org. Lett. 2005, 7, 5163–5165. doi:10.1021/ol051993e |
| 16. | Petrović, M.; Scarpi, D.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E. G. Eur. J. Org. Chem. 2015, 3943–3956. doi:10.1002/ejoc.201500462 |
| 50. | Dharuman, S.; Vankar, Y. D. Org. Lett. 2014, 16, 1172–1175. doi:10.1021/ol500039s |
| 51. | van den Broek, S. B. A. M. W.; Rensen, P. G. W.; van Delft, F. L.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 5906–5912. doi:10.1002/ejoc.201000587 |
| 48. | Larivée, A.; Charette, A. B. Org. Lett. 2006, 8, 3955–3957. doi:10.1021/ol061415d |
| 49. | Jana, S.; Rainier, J. D. Org. Lett. 2013, 15, 4426–4429. doi:10.1021/ol401974v |
| 46. | Norton Matos, M.; Afonso, C. A. M.; Batey, R. A. Tetrahedron 2005, 61, 1221–1244. doi:10.1016/j.tet.2004.11.035 |
| 47. | Zhang, H.; Hay, E. B.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2013, 135, 16610–16617. doi:10.1021/ja408387d |
© 2020 Zanella et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)