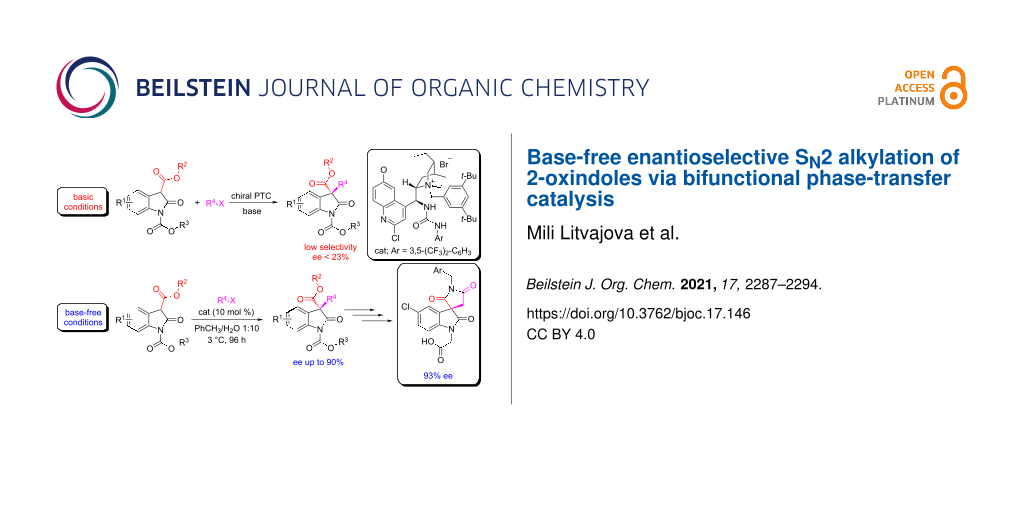
Abstract
N-Protected oxindole derivatives of unprecedented malleability bearing ester moieties at C-3 have been shown to participate in enantioselective phase-transfer-catalysed alkylations promoted by ad-hoc designed quaternary ammonium salts derived from quinine bearing hydrogen-bond donating substituents. For the first time in such phase-transfer-catalysed enolate alkylations, the reactions were carried out under base-free conditions. It was found that urea-based catalysts outperformed squaramide derivatives, and that the installation of a chlorine atom adjacent to the catalyst’s quinoline moiety aided in avoiding selectivity-reducing complications related to the production of HBr in these processes. The influence of steric and electronic factors from both the perspective of the nucleophile and electrophile were investigated and levels of enantiocontrol up to 90% ee obtained. The synthetic utility of the methodology was demonstrated via the concise enantioselective synthesis of a potent CRTH2 receptor antagonist.

Graphical Abstract
Introduction
The 2-oxindole scaffold is an important motif present in a myriad of natural products. Among 2-oxidole derivatives, 3,3'-disubstituted-2-oxindoles are particularly widespread and can also be found in a diverse array of pharmaceutical agents (Figure 1A) [1-4].
![[1860-5397-17-146-1]](/bjoc/content/figures/1860-5397-17-146-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The importance of the 3,3-spirooxindole core and its access through enantioselective enolate alkylation.
Figure 1: The importance of the 3,3-spirooxindole core and its access through enantioselective enolate alkyla...
In addition, their facile transformation into pyrroloindoline and spirooxindole derivatives as well as more structurally complex molecules renders them potentially highly valuable synthetic building blocks [5-12].
Both pyrroloindolines 1 and spirooxindoles 2 are conceivably available from key 3,3-disubstituted intermediates 3, which could be prepared via an enantioselective SN2 alkylation involving enolate 3a (Figure 1B). The versatility of this approach is significantly enhanced when both the substituents at the 3-position are modifiable as much as possible to facilitate further transformations.
In this context, we realised that phase-transfer catalysis, due to its operational simplicity and utility in mediating reactions involving charged intermediates, could be an excellent methodology for the enantioselective SN2 alkylation of enolates derived from the 2-oxindole core [13-23]. In recent years, several examples regarding the alkylation of 3-subsituted-2-oxindoles, via asymmetric phase-transfer catalysis, have been reported [24-30].
However, despite the excellent levels of enantiocontrol often achieved, in the majority of these studies the 2-oxindole subjected to enantioselective alkylation lacks the structural architecture necessary for further modifications (Scheme 1A), presenting instead a fixed – not easily modifiable – group which is not ideal for a modular approach to the construction of more complex molecules such as those shown in Figure 1A. Recently, we partially overcame this challenge by developing a highly enantioselective phase-transfer-catalysed methodology for the SN2 alkylation of methylene ester-substituted 2-oxindole 4 [31]. The utility of this methodology has been demonstrated through the total synthesis of (−)-debromoflustramine B (Scheme 1B). In an attempt to devise variants of this reaction of greater versatility and synthetic utility; we sought to employ the intriguing substrate 5.
![[1860-5397-17-146-i1]](/bjoc/content/inline/1860-5397-17-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A) SN2 alkylation of 3-subtituted-2-oxindoles not readily functionalisable; B) Previous work: enantioselective synthesis of a malleable 2-oxindole capable of further manipulations; C) This work: base-free enantioselective alkylation of 2-oxindoles 5.
Scheme 1: A) SN2 alkylation of 3-subtituted-2-oxindoles not readily functionalisable; B) Previous work: enant...
In comparison with 2-oxindole 4, compound 5 possesses ester functionality directly attached to the 2-oxindole ring – which would provide a functional handle at this position of considerably greater plasticity than anything previously evaluated in the literature. On the other hand, such electron-withdrawing groups, α to the reactive centre, dramatically changes the acidity of the substrate (and thus the reactivity of the enolate conjugate base) and as consequence its reactivity which can drastically impact the enantioselectivity in SN2 alkylation processes. In this report we disclose the outcome of an investigation into the design of an efficient catalytic asymmetric system capable of manipulating this substrate and its application to the enantioselective synthesis of the potent CRTH2 receptor antagonist 6 [32] (Scheme 1C).
Results and Discussion
We began our investigation by evaluating, as a model alkylation, the phase-transfer-catalysed benzylation of substrate 5 under ‘classical’ basic reaction conditions using cinchona alkaloid-based catalysts capable of hydrogen-bonding as a control element [33-40]. As expected, the ester group α to the reactive centre dramatically increases the acidity at this position and, in preliminary studies, we found that under biphasic basic conditions 2-oxindole 5 was undergoing alkylation also in the absence of a phase-transfer catalyst (not ideal when designing a catalytic enantioselective process).
Despite investigating the effects of different solvents, bases and buffer systems, in preliminary experiments we were not able to prevent the non-catalysed benzylation of substrate 5; nevertheless, the enantioselective alkylation of 5 with benzyl bromide in the presence of a phase-transfer catalyst was attempted. This catalytic reaction exhibited poor enantioselectivity and none of the catalysts employed were able to promote the reaction with product ee higher than 22% (Scheme 1C – for more details see Supporting Information File 1).
Over the last decade, Maruoka and co-workers discovered that phase-transfer-catalysed reactions can be occasionally performed even in absence of base under water-enriched/organic biphasic conditions [41-47]. Taking inspiration from these studies, it was envisaged that by employing base-free neutral reaction conditions – given the likely acidity of substrate 5 – that it could be possible to develop an effective catalytic asymmetric protocol.
To the best of our knowledge such base-free catalytic systems have never been applied to processes such as the alkylation of enolates generated in situ. These reactions would produce stoichiometric amounts of acid, which can inhibit the formation of the reactive enolate by driving the enol/enolate equilibrium toward the enol form.
While these considerations seemed discouraging, we were able to define a set of base-free/water-rich reaction conditions suitable for our catalytic system where the formation of the alkylated product was not observed in the absence of a phase-transfer catalyst after a prolonged reaction time of 504 hours (see Supporting Information File 1).
With this new set of conditions in hand, a rational catalyst design process commenced, aimed at improving the selectivity of the base-free SN2 alkylation process.
In preliminary studies, we observed that a substituent at the catalyst C-2' position was enhancing the enantioselectivity of the reaction. Initial attention was therefore focused on the influence the other catalyst subunits (i.e., catalysts 7–9, Table 1) exerted over both reactivity and selectivity.
Attention first turned to the catalyst’s N-substituent. Catalyst 7a, bearing a benzyl group, was able to promote the transformation of 5a in moderate enantioselectivity (Table 1, entry 1). Modification of the N-benzyl unit to incorporate either electron-withdrawing or bulky substituents did not lead to appreciable variations, with the latter leading to a marginal improvement (Table 1, entries 2 and 3). As observed in earlier studies [31], the employment of a N-9-methylantracenyl-substituted catalyst (i.e., 7d) caused a dramatic loss of enantiocontrol (Table 1, entry 4).
Table 1: Catalyst evaluation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-146-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst | loading (mol %) | temp (°C) | time (h) | conv (%)a | ee (%)b |
| 1 | 7a | 5 | rt | 90 | >99 | 50 |
| 2 | 7b | 5 | rt | 114 | 90 | 49 |
| 3 | 7c | 5 | rt | 45 | >99 | 52 |
| 4 | 7d | 5 | rt | 168 | 54 | 7 |
| 5 | 7e | 5 | rt | 48 | >99 | 34 |
| 6 | 7f | 5 | rt | 114 | 97 | 2 |
| 7 | 7g | 5 | rt | 114 | >99 | 0 |
| 8 | 8 | 5 | rt | 161 | 46 | 5 |
| 9 | 9a | 5 | rt | 60 | >99 | 56 |
| 10 | 9b | 5 | rt | 48 | >99 | 55 |
| 11 | 9c | 5 | rt | 45 | >99 | 53 |
| 12 | 7c | 10 | 3 | 144 | >99 | 59 |
| 13 | 9b | 10 | 3 | 144 | >99 | 62 |
| 14 | 9a | 10 | 3 | 144 | >99 | 58 |
| 15 | 9c | 10 | 3 | 144 | >99 | 58 |
aDetermined by 1H NMR spectroscopic analysis using 4-iodoanisole as internal standard. bDetermined by CSP-HPLC.
Modifications to the hydrogen bond-donating functionality – while keeping the N-3,5-di-tert-butylbenzyl unit unchanged – were then introduced. Removing the two electron-withdrawing -CF3 groups from the ureaphenyl moiety resulted in diminished enantioselectivity (Table 1, entry 5), whereas increasing the steric demand in this region of the catalyst led to racemic products (Table 1, entries 6 and 7). Employing a different hydrogen bond-donating motif such as the squaramide (catalyst 8, Table 1, entry 8) resulted in a substantial drop of the enantiocontrol as well as in the reduction of the reaction rate [29,31] – probably due to the ability of squaramides to bind anionic species more strongly than ureas.
The moderate enantiocontrol observed thus far prompted us to posit that the nitrogen atom on the quinoline moiety of the catalyst could participate to the deprotonation of 5a, therefore, leading to less selective alkylation.
In order to test this hypothesis, we designed novel dihydroquinine-derived catalysts of general type 9 bearing an electron-withdrawing substituent at the C-2' position with the intent of lowering the basicity of the quinoline ring. In addition, we prepared a C-2'-phenyl-substituted dihydroquinine-derived catalyst (9c) for comparison. Rather disappointingly, the improvement was marginal (Table 1, entries 9–11) with 9a affording product 10Aa in 56% ee. Therefore, we decided to evaluate the most promising catalysts at lower temperature (3 °C) using 10 mol % catalyst loading. Under these reaction conditions, the chloro derivative 9b proved to be the most efficient catalyst – mediating the formation of product 10Aa in 62% ee after full conversion (Table 1, entries 12–15).
Attention then turned to the 2-oxindole structure. Due to solubility issues chlorobenzene was chosen as the preferred solvent (Figure 2).
![[1860-5397-17-146-2]](/bjoc/content/figures/1860-5397-17-146-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Substrate scope. aIsolated yield. bDetermined by CSP-HPLC. cValue in brackets refers to reaction conducted at 3 °C, using 10 mol % of catalyst. dPerformed in PhCH3. ePerformed using 7c as the catalyst.
Figure 2: Substrate scope. aIsolated yield. bDetermined by CSP-HPLC. cValue in brackets refers to reaction co...
Introduction of a bromo substituent in proximity to the reaction centre led to the formation of product 10Ba in 72% yield with an augmented 83% ee. Disappointingly, 2-oxindoles incorporating similar substituents at different locations, such as the 5-position, afforded products only in moderate ee, with lower enantiocontrol associated with groups possessing greater electron-withdrawing character (i.e., 10Da, 10Ca, 10Ea). Finally, modifications on both the substrate ester and carbamate-moieties did not afford remarkably different outcomes (i.e., 10Fa, 10Ga).
We continued our studies by investigating the behaviour of different alkylating agents (Table 2).
Table 2: Electrophile scope.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-146-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | electrophile | product | time (h) | yields (%) | ee (%)a |
| 1 | a | 10Aa | 144 | 94 | 62 |
| 2 | b | 10Ab | 144 | 94 | 72 |
| 3 | c | 10Ac | 120 | 92 | 84 |
| 4 | d | 10Ad | 144 | 96 | 66 |
| 5b | e | 10Ae | 24 | 94 | 68 |
| 6 | f | 10Af | 144 | 96 | 74 |
| 7 | g | 10Ag | 144 | 97 | 79 |
| 8 | h | 10Ah | 120 | 89 | 80 |
| 9 | i | 10Ai | 144 | 90 | 90 |
| 10b | j | 10Aj | 48 | 99c | 51 |
| 11b | k | 10Ak | 24 | 99c | 54 |
| 12 | l | 10Al | 96 | 98 | 76 |
| 13 | m | 10Am | 96 | 90 | 70 |
aDetermined by CSP-HPLC. bAt rt. cDetermined by 1H NMR spectroscopic analysis using 4-iodoanisole as internal standard.
Reactions with alkylating agents with increased steric demand provided products in higher ee (compare Table 2, entries 1, 2 and 3); with 3,5-bis(tert-butyl)benzyl bromide allowing the isolation of oxindole 10Ac in 92% yield and 84% ee. The employment of benzyl bromides bearing electron-withdrawing groups led to products with moderate ee in the cases of para-substituted analogues (Table 2, entries 4 and 5) while an increase in enantioselectivity, up to 79% ee, was observed using meta-substituted variants (Table 2, entries 6 and 7).
To our delight, relatively electron-rich benzyl bromides were able to afford products in high yields and with improved product ee – up to 90% (Table 2, entries 8 and 9). Attention then switched to non-benzyl bromide-based electrophiles – however, use of allyl iodide was able to furnish product 10Aj with only 51% ee (Table 2, entry 10). Consistent with the goal of developing a protocol of the best possible synthetic utility; alkylating agents which would be easily modified after installation on the oxindole core – such as α-iodoesters – were also evaluated (Table 2, entries 10–12). Although, alkyl esters participated in less enantioselective chemistry (Table 2, entry 11); it was possible to achieve moderate enantiocontrol by employing aromatic ester derivatives, with product 10Al obtained in 98% yield and 76% ee (Table 2, entry 12).
The potential utility of this newly developed methodology was demonstrated through the enantioselective synthesis of the (S)-antipode potent CRTH2 receptor antagonist 6 [48] (Scheme 2).
![[1860-5397-17-146-i2]](/bjoc/content/inline/1860-5397-17-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Enantioselective synthesis of a CRTH2 receptor antagonist.
Scheme 2: Enantioselective synthesis of a CRTH2 receptor antagonist.
Compound 10Al, isolated in 76% ee, was recrystallised in n-hexane to obtain optically pure 10Al. This material was deprotected with benzylamine 11 to afford oxindole 12, which was subsequently N-alkylated with bromo ester 13. The formed product (i.e., 14) was first amidated and then cyclised using benzylamine 11 to generate spirooxindole 15 in 54% yield and 94% ee. Chlorination with NCS, followed by tert-butyl ester cleavage in TFA/CH2Cl2 provided the final bioactive compound 6 in 93% ee.
Conclusion
In conclusion, we have described a base-free protocol for the asymmetric phase-transfer-catalysed SN2 alkylation of densely functionalised 2-oxindole derivatives, employing a biphasic water-rich solvent system. To the best of our knowledge, these base-free neutral reaction conditions have never previously been applied to phase-transfer-catalysed SN2 enolate alkylation reactions and represents an effective process for the generation of carbonaceous quaternary stereocentres.
The process generates malleable di-ester and mono-ester benzylated oxindole substrates which can easily give access to products of biological interest, as evidenced by the facile preparation of the (S)-enantiomer of a potent CRTH2 receptor antagonist.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 10.6 MB | Download |
References
-
Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041
Return to citation in text: [1] -
Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8
Return to citation in text: [1] -
Görmen, M.; Le Goff, R.; Lawson, A. M.; Daïch, A.; Comesse, S. Tetrahedron Lett. 2013, 54, 2174–2176. doi:10.1016/j.tetlet.2013.02.047
Return to citation in text: [1] -
Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Trost, B.; Brennan, M. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161
Return to citation in text: [1] -
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e
Return to citation in text: [1] -
Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/c2ob25184a
Return to citation in text: [1] -
Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808
Return to citation in text: [1] -
Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r
Return to citation in text: [1] -
Dalpozzo, R. Adv. Synth. Catal. 2017, 359, 1772–1810. doi:10.1002/adsc.201700361
Return to citation in text: [1] -
Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e
Return to citation in text: [1] -
O'Donnell, M. J. Acc. Chem. Res. 2004, 37, 506–517. doi:10.1021/ar0300625
Return to citation in text: [1] -
Lygo, B.; Andrews, B. I. Acc. Chem. Res. 2004, 37, 518–525. doi:10.1021/ar030058t
Return to citation in text: [1] -
Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737
Return to citation in text: [1] -
Hashimoto, T.; Maruoka, K. Chem. Rev. 2007, 107, 5656–5682. doi:10.1021/cr068368n
Return to citation in text: [1] -
Jew, S.-s.; Park, H.-g. Chem. Commun. 2009, 7090–7103. doi:10.1039/b914028j
Return to citation in text: [1] -
Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. doi:10.1002/anie.201206835
Return to citation in text: [1] -
Kaneko, S.; Kumatabara, Y.; Shirakawa, S. Org. Biomol. Chem. 2016, 14, 5367–5376. doi:10.1039/c5ob02446c
Return to citation in text: [1] -
Wang, H. Catalysts 2019, 9, 244. doi:10.3390/catal9030244
Return to citation in text: [1] -
Nakamura, T.; Okuno, K.; Nishiyori, R.; Shirakawa, S. Chem. – Asian J. 2020, 15, 463–472. doi:10.1002/asia.201901652
Return to citation in text: [1] -
Majdecki, M.; Niedbała, P.; Tyszka-Gumkowska, A.; Jurczak, J. Synthesis 2021, 53, 2777–2786. doi:10.1055/a-1472-7999
Return to citation in text: [1] -
Lee, T. B. K.; Wong, G. S. K. J. Org. Chem. 1991, 56, 872–875. doi:10.1021/jo00002a074
Return to citation in text: [1] -
Ohmatsu, K.; Kiyokawa, M.; Ooi, T. J. Am. Chem. Soc. 2011, 133, 1307–1309. doi:10.1021/ja1102844
Return to citation in text: [1] -
Chen, W.; Yang, W.; Yan, L.; Tan, C.-H.; Jiang, Z. Chem. Commun. 2013, 49, 9854–9856. doi:10.1039/c3cc46111d
Return to citation in text: [1] -
Ohmatsu, K.; Ando, Y.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 18706–18709. doi:10.1021/ja411647x
Return to citation in text: [1] -
Deng, F.; Moteki, S. A.; Maruoka, K. Asian J. Org. Chem. 2014, 3, 395–398. doi:10.1002/ajoc.201400011
Return to citation in text: [1] -
Sorrentino, E.; Connon, S. J. Org. Lett. 2016, 18, 5204–5207. doi:10.1021/acs.orglett.6b02398
Return to citation in text: [1] [2] -
Ohmatsu, K.; Furukawa, Y.; Kiyokawa, M.; Ooi, T. Chem. Commun. 2017, 53, 13113–13116. doi:10.1039/c7cc07122a
Return to citation in text: [1] -
Craig, R.; Sorrentino, E.; Connon, S. J. Chem. – Eur. J. 2018, 24, 4528–4531. doi:10.1002/chem.201800313
Return to citation in text: [1] [2] [3] -
Crosignani, S.; Page, P.; Missotten, M.; Colovray, V.; Cleva, C.; Arrighi, J.-F.; Atherall, J.; Macritchie, J.; Martin, T.; Humbert, Y.; Gaudet, M.; Pupowicz, D.; Maio, M.; Pittet, P.-A.; Golzio, L.; Giachetti, C.; Rocha, C.; Bernardinelli, G.; Filinchuk, Y.; Scheer, A.; Schwarz, M. K.; Chollet, A. J. Med. Chem. 2008, 51, 2227–2243. doi:10.1021/jm701383e
Return to citation in text: [1] -
Bernal, P.; Fernández, R.; Lassaletta, J. M. Chem. – Eur. J. 2010, 16, 7714–7718. doi:10.1002/chem.201001107
Return to citation in text: [1] -
Johnson, K. M.; Rattley, M. S.; Sladojevich, F.; Barber, D. M.; Nuñez, M. G.; Goldys, A. M.; Dixon, D. J. Org. Lett. 2012, 14, 2492–2495. doi:10.1021/ol300779x
Return to citation in text: [1] -
Li, M.; Woods, P. A.; Smith, M. D. Chem. Sci. 2013, 4, 2907–2911. doi:10.1039/c3sc50592h
Return to citation in text: [1] -
Wang, B.; He, Y.; Fu, X.; Wei, Z.; Lin, Y.; Duan, H. Synlett 2015, 26, 2588–2592. doi:10.1055/s-0035-1560645
Return to citation in text: [1] -
Genoni, A.; Benaglia, M.; Mattiolo, E.; Rossi, S.; Raimondi, L.; Barrulas, P. C.; Burke, A. J. Tetrahedron Lett. 2015, 56, 5752–5756. doi:10.1016/j.tetlet.2015.08.086
Return to citation in text: [1] -
Craig, R.; Litvajova, M.; Cronin, S. A.; Connon, S. J. Chem. Commun. 2018, 54, 10108–10111. doi:10.1039/c8cc05692g
Return to citation in text: [1] -
Lu, N.; Fang, Y.; Gao, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. J. Org. Chem. 2018, 83, 1486–1492. doi:10.1021/acs.joc.7b03078
Return to citation in text: [1] -
Wang, J.; Liu, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. J. Org. Chem. 2020, 85, 4047–4057. doi:10.1021/acs.joc.9b03025
Return to citation in text: [1] -
He, R.; Shirakawa, S.; Maruoka, K. J. Am. Chem. Soc. 2009, 131, 16620–16621. doi:10.1021/ja906821y
Return to citation in text: [1] -
Shirakawa, S.; Kasai, A.; Tokuda, T.; Maruoka, K. Chem. Sci. 2013, 4, 2248–2252. doi:10.1039/c3sc22130j
Return to citation in text: [1] -
Shirakawa, S.; Maruoka, K. Tetrahedron Lett. 2014, 55, 3833–3839. doi:10.1016/j.tetlet.2014.05.014
Return to citation in text: [1] -
Shirakawa, S.; Wang, L.; He, R.; Arimitsu, S.; Maruoka, K. Chem. – Asian J. 2014, 9, 1586–1593. doi:10.1002/asia.201402194
Return to citation in text: [1] -
Kamiyama, T.; Sigrist, L.; Cvengroš, J. Synthesis 2016, 48, 3957–3964. doi:10.1055/s-0035-1562461
Return to citation in text: [1] -
Liu, S.; Maruoka, K.; Shirakawa, S. Angew. Chem., Int. Ed. 2017, 56, 4819–4823. doi:10.1002/anie.201612328
Return to citation in text: [1] -
Zhu, J.; Cui, D.; Li, Y.; He, J.; Chen, W.; Wang, P. Org. Biomol. Chem. 2018, 16, 3012–3017. doi:10.1039/c8ob00583d
Return to citation in text: [1] -
We identified 6 as the S enantiomer. The absolute configuration was assigned by analogy to the absolute configuration of 10Al, which was determined using XRD analysis (structure submitted to the CCDC No. 2089007).
Return to citation in text: [1]
| 1. | Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041 |
| 2. | Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8 |
| 3. | Görmen, M.; Le Goff, R.; Lawson, A. M.; Daïch, A.; Comesse, S. Tetrahedron Lett. 2013, 54, 2174–2176. doi:10.1016/j.tetlet.2013.02.047 |
| 4. | Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056 |
| 31. | Craig, R.; Sorrentino, E.; Connon, S. J. Chem. – Eur. J. 2018, 24, 4528–4531. doi:10.1002/chem.201800313 |
| 24. | Lee, T. B. K.; Wong, G. S. K. J. Org. Chem. 1991, 56, 872–875. doi:10.1021/jo00002a074 |
| 25. | Ohmatsu, K.; Kiyokawa, M.; Ooi, T. J. Am. Chem. Soc. 2011, 133, 1307–1309. doi:10.1021/ja1102844 |
| 26. | Chen, W.; Yang, W.; Yan, L.; Tan, C.-H.; Jiang, Z. Chem. Commun. 2013, 49, 9854–9856. doi:10.1039/c3cc46111d |
| 27. | Ohmatsu, K.; Ando, Y.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 18706–18709. doi:10.1021/ja411647x |
| 28. | Deng, F.; Moteki, S. A.; Maruoka, K. Asian J. Org. Chem. 2014, 3, 395–398. doi:10.1002/ajoc.201400011 |
| 29. | Sorrentino, E.; Connon, S. J. Org. Lett. 2016, 18, 5204–5207. doi:10.1021/acs.orglett.6b02398 |
| 30. | Ohmatsu, K.; Furukawa, Y.; Kiyokawa, M.; Ooi, T. Chem. Commun. 2017, 53, 13113–13116. doi:10.1039/c7cc07122a |
| 13. | Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e |
| 14. | O'Donnell, M. J. Acc. Chem. Res. 2004, 37, 506–517. doi:10.1021/ar0300625 |
| 15. | Lygo, B.; Andrews, B. I. Acc. Chem. Res. 2004, 37, 518–525. doi:10.1021/ar030058t |
| 16. | Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737 |
| 17. | Hashimoto, T.; Maruoka, K. Chem. Rev. 2007, 107, 5656–5682. doi:10.1021/cr068368n |
| 18. | Jew, S.-s.; Park, H.-g. Chem. Commun. 2009, 7090–7103. doi:10.1039/b914028j |
| 19. | Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. doi:10.1002/anie.201206835 |
| 20. | Kaneko, S.; Kumatabara, Y.; Shirakawa, S. Org. Biomol. Chem. 2016, 14, 5367–5376. doi:10.1039/c5ob02446c |
| 21. | Wang, H. Catalysts 2019, 9, 244. doi:10.3390/catal9030244 |
| 22. | Nakamura, T.; Okuno, K.; Nishiyori, R.; Shirakawa, S. Chem. – Asian J. 2020, 15, 463–472. doi:10.1002/asia.201901652 |
| 23. | Majdecki, M.; Niedbała, P.; Tyszka-Gumkowska, A.; Jurczak, J. Synthesis 2021, 53, 2777–2786. doi:10.1055/a-1472-7999 |
| 5. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 6. | Trost, B.; Brennan, M. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975 |
| 7. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161 |
| 8. | Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247–7290. doi:10.1039/c2cs35100e |
| 9. | Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/c2ob25184a |
| 10. | Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808 |
| 11. | Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r |
| 12. | Dalpozzo, R. Adv. Synth. Catal. 2017, 359, 1772–1810. doi:10.1002/adsc.201700361 |
| 31. | Craig, R.; Sorrentino, E.; Connon, S. J. Chem. – Eur. J. 2018, 24, 4528–4531. doi:10.1002/chem.201800313 |
| 48. | We identified 6 as the S enantiomer. The absolute configuration was assigned by analogy to the absolute configuration of 10Al, which was determined using XRD analysis (structure submitted to the CCDC No. 2089007). |
| 41. | He, R.; Shirakawa, S.; Maruoka, K. J. Am. Chem. Soc. 2009, 131, 16620–16621. doi:10.1021/ja906821y |
| 42. | Shirakawa, S.; Kasai, A.; Tokuda, T.; Maruoka, K. Chem. Sci. 2013, 4, 2248–2252. doi:10.1039/c3sc22130j |
| 43. | Shirakawa, S.; Maruoka, K. Tetrahedron Lett. 2014, 55, 3833–3839. doi:10.1016/j.tetlet.2014.05.014 |
| 44. | Shirakawa, S.; Wang, L.; He, R.; Arimitsu, S.; Maruoka, K. Chem. – Asian J. 2014, 9, 1586–1593. doi:10.1002/asia.201402194 |
| 45. | Kamiyama, T.; Sigrist, L.; Cvengroš, J. Synthesis 2016, 48, 3957–3964. doi:10.1055/s-0035-1562461 |
| 46. | Liu, S.; Maruoka, K.; Shirakawa, S. Angew. Chem., Int. Ed. 2017, 56, 4819–4823. doi:10.1002/anie.201612328 |
| 47. | Zhu, J.; Cui, D.; Li, Y.; He, J.; Chen, W.; Wang, P. Org. Biomol. Chem. 2018, 16, 3012–3017. doi:10.1039/c8ob00583d |
| 33. | Bernal, P.; Fernández, R.; Lassaletta, J. M. Chem. – Eur. J. 2010, 16, 7714–7718. doi:10.1002/chem.201001107 |
| 34. | Johnson, K. M.; Rattley, M. S.; Sladojevich, F.; Barber, D. M.; Nuñez, M. G.; Goldys, A. M.; Dixon, D. J. Org. Lett. 2012, 14, 2492–2495. doi:10.1021/ol300779x |
| 35. | Li, M.; Woods, P. A.; Smith, M. D. Chem. Sci. 2013, 4, 2907–2911. doi:10.1039/c3sc50592h |
| 36. | Wang, B.; He, Y.; Fu, X.; Wei, Z.; Lin, Y.; Duan, H. Synlett 2015, 26, 2588–2592. doi:10.1055/s-0035-1560645 |
| 37. | Genoni, A.; Benaglia, M.; Mattiolo, E.; Rossi, S.; Raimondi, L.; Barrulas, P. C.; Burke, A. J. Tetrahedron Lett. 2015, 56, 5752–5756. doi:10.1016/j.tetlet.2015.08.086 |
| 38. | Craig, R.; Litvajova, M.; Cronin, S. A.; Connon, S. J. Chem. Commun. 2018, 54, 10108–10111. doi:10.1039/c8cc05692g |
| 39. | Lu, N.; Fang, Y.; Gao, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. J. Org. Chem. 2018, 83, 1486–1492. doi:10.1021/acs.joc.7b03078 |
| 40. | Wang, J.; Liu, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. J. Org. Chem. 2020, 85, 4047–4057. doi:10.1021/acs.joc.9b03025 |
| 32. | Crosignani, S.; Page, P.; Missotten, M.; Colovray, V.; Cleva, C.; Arrighi, J.-F.; Atherall, J.; Macritchie, J.; Martin, T.; Humbert, Y.; Gaudet, M.; Pupowicz, D.; Maio, M.; Pittet, P.-A.; Golzio, L.; Giachetti, C.; Rocha, C.; Bernardinelli, G.; Filinchuk, Y.; Scheer, A.; Schwarz, M. K.; Chollet, A. J. Med. Chem. 2008, 51, 2227–2243. doi:10.1021/jm701383e |
| 29. | Sorrentino, E.; Connon, S. J. Org. Lett. 2016, 18, 5204–5207. doi:10.1021/acs.orglett.6b02398 |
| 31. | Craig, R.; Sorrentino, E.; Connon, S. J. Chem. – Eur. J. 2018, 24, 4528–4531. doi:10.1002/chem.201800313 |
© 2021 Litvajova et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)