Abstract
A genome mining survey combined with metabolome analysis of publicly available strains identified Couchioplanes sp. RD010705, a strain belonging to an underexplored genus of rare actinomycetes, as a producer of new metabolites. HPLC-DAD-guided fractionation of its fermentation extracts resulted in the isolation of five new methyl-branched unsaturated fatty acids, (2E,4E)-2,4-dimethyl-2,4-octadienoic acid (1), (2E,4E)-2,4,7-trimethyl-2,4-octadienoic acid (2), (R)-(−)-phialomustin B (3), (2E,4E)-7-hydroxy-2,4-dimethyl-2,4-octadienoic acid (4), (2E,4E)-7-hydroxy-2,4,7-trimethyl-2,4-octadienoic acid (5), and one prenylated tryptophan derivative, 6-(3,3-dimethylallyl)-N-acetyl-ʟ-tryptophan (6). The enantiomer ratio of 4 was determined to be approximately S/R = 56:44 by a recursive application of Trost’s chiral anisotropy analysis and chiral HPLC analysis of its methyl ester. Compounds 1–5 were weakly inhibitory against Kocuria rhizophila at MIC 100 μg/mL and none were cytotoxic against P388 at the same concentration.

Graphical Abstract
Introduction
Actinomycetes, a subgroup of filamentous Gram-positive bacteria within the class Actinomycetales, have provided many important clinical drugs [1,2], agrochemicals [3], food additives [4,5], and biochemical reagents [6-9], and continue to be a core source of bioactive molecules [10]. While most of the actinomycetes-derived compounds have been reported from the genus Streptomyces [11], non-Streptomyces actinomycetes, commonly referred to as rare actinomycetes [12], are attracting considerable attention as less tapped taxa for drug discovery. Those within the family Micromonosporaceae, represented by the second most prolific genus Micromonospora following Streptomyces, are especially noted, accounting for more than 800 metabolites of actinomycetes origin [11], which include the antiinfective aminoglycoside gentamicin [13], antidiabetic glycoside acarbose [14], glycopeptide antibiotic teicoplanin [15], enediyne antitumor component of drug-antibody conjugate calicheamicin γ1I [16], and Clostridium difficile-specific macrolide fidaxomicin [17].
At present, only 5 out of 29 valid genera in this family [18] – Actinoplanes, Dactylosporangium, Micromonospora, Salinispora, and Verrucosispora – have mainly been investigated [11] and the remaining 24 are still untouched or underexplored. However, in silico genome mining identified multiple secondary metabolite biosynthetic gene clusters in selected strains from minor actinomycetes genera, implying their comparable biosynthetic capacities to those of the already proven genera [19]. Encouraged by these reports, we examined the metabolites of Pseudosporangium sp. RD062863, a strain available at the culture collection of the Biological Resource Center, National Institute of Technology and Evaluation (NBRC) [20], and discovered a novel cyclopeptide pseudosporamide along with three new oligomycin-class polyketide [21]. In addition, the same approach to the different family (Pseudonocardiaceae) yielded mycetoindole, a new class of dehydrotryptophan derivative from Actinomycetospora [22].
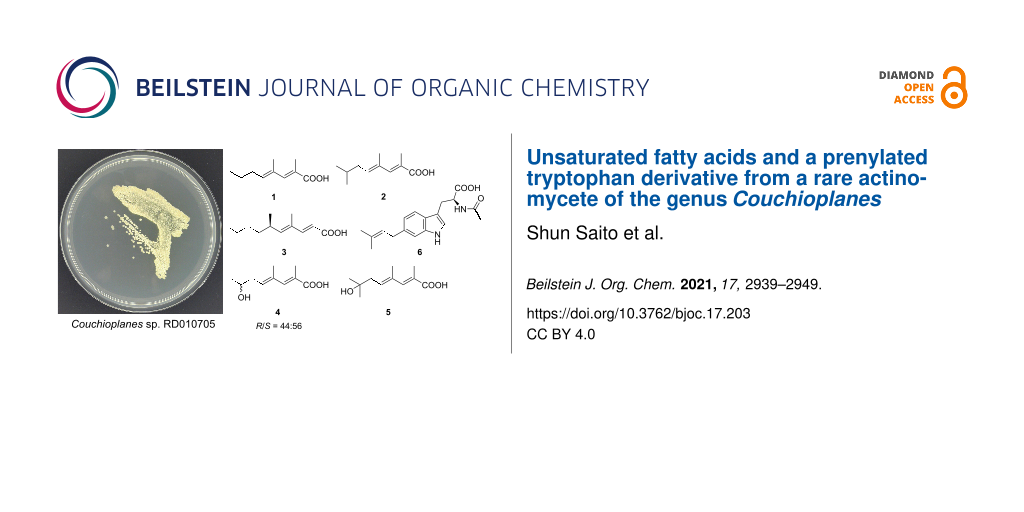
As part of our continuing studies on the metabolites from underexplored rare actinomycetes, the genus Couchioplanes, another minor genus in Micromonosporaceae first isolated in 1994 from a sandy soil in Japan [23], was set to be the next target. While the anti-SMASH-assisted genome mining [24] in C. caeruleus DSM 43634 revealed approximately 20 secondary metabolite biosynthetic gene clusters, only one compound, heptaene macrolide 67-121C, is known to date [25], leaving room for exploration. Four strains of the same genus, available at the NBRC’s culture collection [20], were fermented and their metabolites were analyzed by HPLC-DAD, which detected several prominent peaks from the culture extracts of strain RD010705. Fractionation and purification guided by characteristic UV spectra led to the discovery of five new methyl-branched unsaturated fatty acids, (2E,4E)-2,4-dimethyl-2,4-octadienoic acid (1), (2E,4E)-2,4,7-trimethyl-2,4-octadienoic acid (2), (R)-(−)-phialomustin B (3), (2E,4E)-7-hydroxy-2,4-dimethyl-2,4-octadienoic acid (4), and (2E,4E)-7-hydroxy-2,4,7-trimethyl-2,4-octadienoic acid (5), as well as one new prenylated tryptophan derivative, 6-(3,3-dimethylallyl)-N-acetyl-ʟ-tryptophan (6) (Figure 1).
![[1860-5397-17-203-1]](/bjoc/content/figures/1860-5397-17-203-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
The producing strain RD010705 was shake-cultured in A16 liquid medium at 30 °C for 8 days, and the whole culture was extracted with 1-butanol. The extract (4.4 g from 3 L) was sequentially fractionated by column chromatographies on silica gel and ODS, and the resulting fractions were purified by reverse-phase HPLC to give 1 (5.2 mg), 2 (2.3 mg), 3 (1.0 mg), 4 (6.3 mg), and 5 (8.0 mg).
The molecular formula of 1 was determined to be C10H16O2 on the basis of its NMR and HR–ESI–TOFMS data (m/z 191.1044 [M + Na]+, Δ + 0.1 mmu). Three degrees of unsaturation, calculated from the molecular formula, a UV absorption maximum at 264 nm, and IR absorption bands at 1679 and 2800–3200 cm−1, suggested dienone and hydroxy functionalities. The 1H, 13C, and HSQC data allowed to assign ten carbon signals to one carbonyl carbon (δC 174.0), two each of non-protonated sp2 carbons (δC 132.5, 123.9), sp2 methines (δC 145.7, 138.5), and sp3 methylenes, and three methyls (Table 1 and Table 2). A sequence of COSY correlations was detected from a triplet methyl H-8 to an olefinic methine H-5 via two methylenes H-7 and H-6, which revealed a propyl-substituted olefin moiety (Figure 2). In addition, HMBC correlations (Table 2) from the allylic methyl H-10 to C-5, C-4, and C-3 and from the other allylic methyl H-9 to C-3, C-2, and C-1 elucidated the carbon connectivity from C-5 to C-1, thus completing the planar structure. The double bond geometries were determined to be both E based on NOESY correlations between H-3 and H-5 and between H-9 and H-10 (Figure 2). Therefore, 1 was determined to be (2E,4E)-2,4-dimethyl-2,4-octadienoic acid.
Table 1: 13C NMR data for 1–5 in CDCl3.
| 1 | 2 | 3 | 4 | 5 | |
| No. | δC, typea | δC, typea | δC, typea | δC, typea | δC, typea |
| 1 | 174.0, C | 173.9, C | 177.1, C | 174.1, C | 174.1, C |
| 2 | 123.9, C | 124.0, C | 115.1, CH | 124.8, C | 124.8, C |
| 3 | 145.7, CH | 145.7, CH | 151.7, CH | 144.8, CH | 145.2, CH |
| 4 | 132.5, C | 132.9, C | 131.5, C | 134.7, C | 134.9, C |
| 5 | 138.5, CH | 137.4, CH | 149.3, CH | 133.0, CH | 132.8, CH |
| 6 | 30.7, CH2 | 37.7, CH2 | 33.3, CH | 38.2, CH2 | 42.5, CH2 |
| 7 | 22.5, CH2 | 28.8, CH | 37.0, CH2 | 67.8, CH | 71.7, C |
| 8 | 14.0, CH3 | 22.6, CH3b | 29.8, CH2 | 23.1, CH3 | 29.4, CH3b |
| 9 | 13.8, CH3 | 22.6, CH3b | 22.9, CH2 | 13.7, CH3 | 29.4, CH3b |
| 10 | 16.3, CH3 | 13.9, CH3 | 14.2, CH3 | 16.5, CH3 | 13.9, CH3 |
| 11 | 16.4, CH3 | 12.4, CH3 | 16.6, CH3 | ||
| 12 | 20.6, CH3 | ||||
aRecorded at 125 MHz (reference δC 77.2). bOverlapping signals.
Table 2: 1H NMR data for 1–5 in CDCl3.
| 1 | 2 | 3 | 4 | 5 | |
| No. | δH, mult (J in Hz)a | δH, mult (J in Hz)a | δH, mult (J in Hz)a | δH, mult (J in Hz)a | δH, mult (J in Hz)a |
| 1 | |||||
| 2 | 5.79, d (15.2) | ||||
| 3 | 7.26, s | 7.26, s | 7.35, d (14.7) | 7.26, s | 7.28, s |
| 4 | |||||
| 5 | 5.70, t (7.2) | 5.72, t (7.5) | 5.68, d (9.1) | 5.72, t (7.3) | 5.79, t (7.6) |
| 6 | 2.14, q (7.5) | 2.05, t (7.1) | 2.51, m | 2.35, m | 2.36, d (7.9) |
| 7 | 1.45, sex (7.4) | 1.71, m | 1.27, mb | 3.94, sex (6.2) | |
| 1.36, m | |||||
| 8 | 0.94, t (7.4) | 0.93, d (6.7)b | 1.21–1.26, m | 1.24, dd (6.2) | 1.27, sb |
| 9 | 2.02, s | 0.93, d (6.7)b | 1.23–1.27, mb | 2.02, s | 1.27, sb |
| 10 | 1.86, s | 2.03, s | 0.87, t (7.3) | 1.89, s | 2.01, s |
| 11 | 1.86, s | 1.77, s | 1.86, s | ||
| 12 | 0.97, d (6.6) | ||||
| No. | HMBCc | HMBCc | HMBCc | HMBCc | HMBCc |
| 1 | |||||
| 2 | |||||
| 3 | 1, 2, 5, 9, 10 | 1, 2, 5, 10, 11 | 1, 2, 5, 9, 10 | 1, 2, 5, 10, 11 | |
| 4 | |||||
| 5 | 3, 6, 7, 10 | 3, 6, 7, 11 | 3, 6, 7, 10 | 3, 6, 7, 11 | |
| 6 | 4, 5, 7, 8 | 4, 5, 7, 8, 9 | 4, 5, 7, 8 | 4, 5, 7, 8, 9 | |
| 7 | 5, 6, 8 | 5, 6, 8, 9 | 8, 9 | 5, 6, 8 | |
| 8 | 6, 7 | 6, 7, 9 | 7, 9 | 6, 7 | 6, 7, 9 |
| 9 | 1, 2, 3 | 6, 7, 8 | 8, 10 | 1, 2, 3 | 6, 7, 8 |
| 10 | 3, 4, 5 | 1, 2, 3 | 8, 9 | 3, 4, 5 | 1, 2, 3 |
| 11 | 3, 4, 5 | 3, 4, 5 | 3, 4, 5 | ||
| 12 | 5, 6, 7 | ||||
aRecorded at 500 MHz (reference δH 7.26). bOverlapping signals. cFrom proton to indicated carbon(s).
![[1860-5397-17-203-2]](/bjoc/content/figures/1860-5397-17-203-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: COSY, key HMBC and NOESY correlations of 1–5.
Figure 2: COSY, key HMBC and NOESY correlations of 1–5.
The 1H NMR spectrum of 2 was similar to those of 1 in overall (Table 1 and Table 2). One major difference was a replacement of the triplet methyl peak in 1 by a six proton-equivalent doublet methyl peak, which suggested an isopropyl-terminated structure. This was proven by a COSY correlation between this methyl proton (H-8/H-9) and a methine proton (H-7) and HMBC correlations from H-8/H-9 to the methine carbon (C-7) and the allylic methylene carbon (C-6). The deduced structure was consistent with a molecular formula C11H18O2 established by HR–ESI–TOFMS analysis (m/z 205.1202 [M + Na]+, Δ + 0.3 mmu), which is larger than 1 by one methylene. NOESY correlations surrounding the double bonds was the same to those observed for 1 (Figure 2), thus concluding 2 to be (2E,4E)-2,4,7-trimethyl-2,4-octadienoic acid.
Compound 3 has a molecular formula of C12H20O2 (m/z 219.1356 [M + Na]+, Δ 0.0 mmu), which is larger by C2H4 than 1. Analysis of a COSY spectrum identified a 1,2-disubstituted (E)-olefin fragment H-2/H-3 (3JH-H = 15.0 Hz), a four-carbon fragment containing a methyl-substituted olefinic methine H-5/H-6/(H-12)/H-7, and an ethyl group H-9/H-10 (Figure 2, Table 1 and Table 2). The connectivity of the former two fragments were intervened by an sp2 quaternary carbon (C-4) substituted by an allylic methyl group (H-11) based on HMBC correlations from H-11 to C-3, C-4, and C-5. Another intervention by a methylene unit (C-8) to connect the second and third fragments was supported by HMBC correlations from H-7 to C-9, H-8 to C-6, and H-10 to C-8, thus completing an alkyl chain part. The remaining atomic composition was CHO2, and despite the lack of evidentiary HMBC correlations, placing a carboxylic acid functionality at the open end (C-2) was reasonable in consideration of the chemical shift of the unused carbon (δH 177.1, C-1) and the molecular formula. A NOESY correlation between H-11 and H-12 supported an E-configuration for the C-4/C-5 double bond. The established planar structure was identical to that of a fungal metabolite phialomustin B [26], for which specific rotation, enumerated 1H and 13C NMR data, and high-resolution ESIMS data were presented. While an S-configuration was assigned for phialomustin B based on its positive specific rotation ([α]D25 +55.5, c 1.5, CHCl3) in comparison with those of synthetic standards [26], opposite negative signs in CHCl3 and MeOH ([α]D27 –12, c 0.035, CHCl3; [α]D25 –76.0, c 0.05, MeOH), though the rotatory power in CHCl3 not as large as expected presumably due to the sample scarcity, supported an R-configuration. Therefore, 3 was speculated to be (R)-(−)-phialomustin B.
The molecular formula of 4, determined to be C10H16O3 on the basis of HR-ESITOF-MS data (m/z 207.0992 [M + Na]+, Δ 0.0 mmu), was one-oxygen larger than that of 1. The 1H NMR spectrum was mostly similar to those of 1 (Table 1 and Table 2), except for the presence of an oxymethine resonance (δH 3.96/δC 67.8) in place of the shielded methylene at C-7, which suggested hydroxylation on the same carbon. This was supported by COSY correlations establishing the connectivity from H-5 to H-8, and completely the same HMBC and NOESY correlations for the remaining part to those observed for 1 and 2 (Figure 2). To address the absolute configuration, 4 was esterified with TMS-diazomethane and the resulting methyl ester 4' was acylated with (R)- or (S)-α-methoxyphenylacetic acid (MPA). To our surprise, however, 1H NMR spectra of both the acylation products were substantially the same but contained several duplicated signals with contrasting peak intensities, indicating 4 to be an enantiomeric mixture of unequal quantities (Figure 3). Indeed, two 1H resonance sets with slightly different chemical shifts were obtained, which were assigned to be a pair of MPA-acylated diastereomers (4'a and 4'b; 4'c and 4'd) by careful interpretation of a COSY spectrum of the (R)-MPA acylation product mixture (Supporting Information File 1, Figure S25). Furthermore, the slightly excessive 4'a and 4'c were identified to be MPA derivatives of (S)-4, and hence less excessive 4'b and 4'd to be those of (R)-4, by recursive application of Trost’s chiral anisotropy rule to the 1H chemical shift differences between the diastereomer pairs. The enantiomer ratio of 4 was estimated to be S/R = 56:44 or near by chiral phase HPLC analysis of 4' on a cellulose tribenzoate-coated silica gel column (Figure 4). Thus, 4 was concluded to be an enantiomeric mixture of (2E,4E)-7-hydroxy-2,4-dimethyl-2,4-octadienoic acid with an approximate enantiomer ratio of S/R = 56:44.
![[1860-5397-17-203-3]](/bjoc/content/figures/1860-5397-17-203-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra and partial chemical shift assignments for MPA esters 4'a–4'd. Upper: (R)-MPA acylation products, Lower: (S)-MPA acylation products.
Figure 3: 1H NMR spectra and partial chemical shift assignments for MPA esters 4'a–4'd. Upper: (R)-MPA acylat...
![[1860-5397-17-203-4]](/bjoc/content/figures/1860-5397-17-203-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Chiral phase HPLC analysis of methyl ester 4'. Italicized numbers indicate peak areas.
Figure 4: Chiral phase HPLC analysis of methyl ester 4'. Italicized numbers indicate peak areas.
The 1H and 13C NMR spectra of 5 were quite similar to those of 4 (Table 1 and Table 2), except for the lack of the oxymethine proton resonance and replacement of the doublet methyl resonance by a singlet signal with a six-proton intensity. These differences, together with a one methylene-larger molecular formula (C11H18O3), suggested a methyl substitution at the carbinol carbon (δC 71.7, C-7). This was verified by HMBC correlations from the singlet methyl proton (H-8/H-9) to the carbinol carbon. Thus, 5 was identified to be (2E,4E)-7-hydroxy-2,4,7-trimethyl-2,4-octadienoic acid.
The same strain also produced another new compound, 6-(3,3-dimethylallyl)-N-acetyl-ʟ-tryptophan (6), when fermented in modified V22, a different medium. This compound was isolated by the same sequence of chromatographies as above, yielding 45 mg from a 6 L culture. The molecular formula was determined to be C18H22N2O3 based on its NMR and HR–ESI–TOFMS data (m/z 313.1556 [M – H]–, Δ – 0.2 mmu), corresponding to nine degrees of unsaturation. The UV spectrum, exhibiting the absorption maxima at 229 and 282 nm, was typical of an indole functionality. The IR absorption bands at 3310 and 1656 cm–1 suggested the presence of OH/NH and carbonyl groups. 1H, 13C, and HSQC spectra (Table 3) revealed the composition of this molecule to be two shielded carbonyls, five other sp2 nonprotonated carbons, five sp2 methines, one sp3 methine, two sp3 methylenes, three singlet methyls, and two amino protons. The remaining OH group should be a part of a carboxylic acid functionality considering the lack of oxygenated carbons besides carbonyls, and two degrees of unsaturation, not accounted for by double bonds, were consistent with the indole ring.
Table 3: NMR data for 6 in CDCl3.
| No. | δC,a type | δH, mult (J in Hz)b | HMBCc |
| 1 | NH | 8.41, s | 2, 3, 3a, 7a |
| 2 | 122.9, CH | 6.91, s | 3, 3a, 7a |
| 3 | 109.9, C | ||
| 3a | 126.3, C | ||
| 4 | 118.6, CH | 7.45, d (8.2) | 3, 3a, 6 |
| 5 | 120.8, CH | 6.92, d (8.3) | 3a, 7, 1' |
| 6 | 136.0, C | ||
| 7 | 110.7, CH | 7.11, s | 3a, 5, 6, 1' |
| 7a | 136.7, C | ||
| 8 | 27.4, CH2 | 3.29, m | 2, 3, 3a, 9, 10 |
| 9 | 53.4, CH | 4.87, m | 3, 8, 10, 11 |
| 10 | 174.3, C | ||
| 11 | NH | 6.26, d (6.9) | 9, 10, 12 |
| 12 | 170.7, C | ||
| 13 | 23.2, CH3 | 1.91, s | 12 |
| 1' | 34.7, CH2 | 3.41, d (6.9) | 5, 6, 7, 2', 3' |
| 2' | 124.1, CH | 5.35, t (7.5) | 1', 4', 5' |
| 3' | 132.1, C | ||
| 4' | 18.0, CH3 | 1.73, sd | 2', 3', 5' |
| 5' | 25.9, CH3 | 1.73, sd | 2', 3', 4' |
aRecorded at 125 MHz (reference δC 77.2). bRecorded at 500 MHz (reference δH 7.26). cFrom proton to indicated carbon(s). dOverlapping signals.
As expected, assembling the above components by COSY and HMBC correlations established 6-prenylated N-acetyltryptophan (Figure 5). The N-acetylation was evident from HMBC correlations from the amide (NH-11) and acetyl methyl protons (H-13) to the amide carbon (C-12), while prenylation at C-6 was supported by HMBC correlations from H-5 and H-7 to C-1' and from H-1' to C-6. The absolute configuration was determined by chiral anisotropy analysis after derivatization with each of the phenylglycine methyl ester (PGME) enantiomers [27], which gave positive ΔδH(S-R) values for NH-1, H-2, H-4, H-5 and H-8 and negative values for H-9, NH-11 and H-13 (Figure 6). Thus, an S-configuration, corresponding to an ʟ-chirality for tryptophan, was assigned.
![[1860-5397-17-203-5]](/bjoc/content/figures/1860-5397-17-203-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: COSY and key HMBC correlations observed for 6.
Figure 5: COSY and key HMBC correlations observed for 6.
![[1860-5397-17-203-6]](/bjoc/content/figures/1860-5397-17-203-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: ΔδH(S-R) values in ppm calculated from PGME amides 6a and 6b.
Figure 6: ΔδH(S-R) values in ppm calculated from PGME amides 6a and 6b.
Compounds 1–6 were not cytotoxic against P388 murine leukemia cells (IC50 > 100 μM), nor antimicrobial against five bacteria, Bacillus subtilis, Staphylococcus aureus, Ralstonia solanacearum, Rhizobium radiobacter and Escherichia coli, or a yeast Candida albicans (MIC > 100 μg/mL). Compounds 1–5 were marginally active against Kocuria rhizophila (MIC = 100 μg/mL).
Conclusion
In this study, five unsaturated fatty acids (1–5) and one prenylated tryptophan derivative (6) were isolated as new natural products from Couchioplanes sp. RD010705. The α,γ-dimethyl-α,γ-dienoyl C8 motif in 1, 2, 4, and 5 is only precedented by 64p-B (2,4-dimethyl-2,4-octadienamide) produced by manumycin-producing Streptomyces, though the physicochemical properties of which is yet to be disclosed [28]. The γ,ε-dimethyl-α,γ-dienoyl motif in 3 is seen in many antibiotics but that with a C10 chain length is only precedented by manumycin C [29], TMC-1 C [30], and phialomustins [26] (Figure 7). In the family Micromonosporaceae, a strain belonging to the genus Plantactinospora is known to produce U-62162, a manumycin-type metabolite with a methyl-branched C9 unsaturated acyl chain [31]. Moreover, salinipyrones, produced by a Salinispora strain, were shown to be biosynthetic byproducts of the rosamicin polyketide synthase [32]. Though not a result from Micromonosporaceae, another example of truncated polyketides is citreodiol, a similarly methyl-branched unsaturated fatty acid ester, which is produced by type I polyketide synthase in a Streptomyces strain by a heterologous expression experiment [33]. These facts suggest that 1–5 could be byproducts from the biosynthesis of larger polyketides, but further investigation is necessary for their biosynthesis. Prenylated indoles are widely distributed among bacteria, fungi and plants, and all seven positions are subject of prenylation except for the bridgehead carbons [34]. Compound 6 is the acetylated derivative of 6-(3,3-dimethylallyl)-ʟ-tryptophan from Streptomyces sp. SN-593 [35]. Further chemical exploration on the genus Couchioplanes will disclose its actual biosynthetic capacity in secondary metabolism.
![[1860-5397-17-203-7]](/bjoc/content/figures/1860-5397-17-203-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Selected examples of the related compounds derived from the strains in the genus Streptomyces (a) and the family Micromonosporaceae (b).
Figure 7: Selected examples of the related compounds derived from the strains in the genus Streptomyces (a) a...
Experimental
General experimental procedures
Optical rotations were measured using a JASCO P-1030 polarimeter. UV spectra were recorded on a Shimadzu UV-1800 UV–vis spectrophotometer. IR spectra were measured on a PerkinElmer Spectrum 100. NMR spectra were obtained on a Bruker AVANCE II 500 or AVANCE NEO 500 spectrometer in DMSO-d6 or CDCl3, and referenced to the residual solvent signals (δH 2.50, δC 39.5 for DMSO-d6; δH 7.26, δC 77.2 for CDCl3). HR–ESI–TOFMS spectra were recorded on a Bruker micrOTOF spectrometer. Silica gel 60 (spherical) (Kanto Chemical Co., Inc.) was used for silica gel column chromatography. Cosmosil 75C18-PREP (Nacalai Tesque, Inc.) was used for ODS column chromatography. Routine HPLC separations were performed on an Agilent HP1200 system, and chiral HPLC analysis was done on a Shimadzu Prominence UFLC system composed of a DGU-20A3R degasser, an SPD-20A UV detector, and an LC-20AD pump, which is connected to a C-R8A Chromatopac data processor.
Microorganism
Strain RD010705 was obtained from NBRC. The strain was identified as a member of the genus Couchioplanes on the basis of 98.2% similarity in the 16S rRNA gene sequence (1418 nucleotides; DDBJ accession number LC512746) to Couchioplanes caeruleus strain DSM 44103T (accession number NR_026295.1).
Fermentation
Strain RD010705, grown on half-strength ISP medium 2 consisting of yeast extract 0.2%, malt extract 0.5%, and glucose 0.2% (pH 7.3) solidified by agar 2%, was inoculated into test tubes (inner diameter, 15 mm; length 16.5 cm) each containing 5 mL of YG seed medium consisting of glucose 1% and yeast extract 1% (pH 7.0). The tubes were shaken at 260 strokes/min at 28 °C for 3 days (TC-500R, Takasaki Scientific Instruments Corp.). One-mL aliquots of the resulting seed cultures were transferred into 500 mL Erlenmeyer flasks each containing 100 mL of production medium and the flasks were fermented on a rotary shaker (TB98, Takasaki Scientific Instruments Corp.) operated at 120 rpm at 30 °C for 8 days.
Isolation of compounds 1–5
Compounds 1–5 were obtained from a culture fermented in A16 production medium with a composition of glucose 2%, Pharmamedia® (Traders Protein, Memphis, TN, USA) 1%, and CaCO3 0.5%. The pH of the medium was adjusted to 7.0 before autoclaving. At the end of fermentation, 100 mL of 1-butanol was added to each flask, and the flasks were allowed to shake for 1 h. The mixture was centrifuged at 6,000 rpm for 10 min and the organic layer was separated from the aqueous layer containing the mycelium. The organic layer was concentrated in vacuo to give 4.4 g of an extract from a 3 L culture. The extract was subjected to silica gel column chromatography eluted with a step gradient of CHCl3/MeOH mixture solvent (1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1 v/v). The sixth fraction eluted with CHCl3/MeOH (1:1) was evaporated to dryness and the oily residue (429 mg) was chromatographed on ODS eluted with a step gradient of MeCN/0.1% HCO2H solutions (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2 v/v). The second, third, and fifth fractions were purified by HPLC on an Nacalai Tesque Cosmosil 5C18-AR-II packed column (10 × 250 mm) eluted with linear gradient elution of MeCN from 15 to 85% over 35 min in 0.1% HCO2H at 4 mL/min to give (2E,4E)-7-hydroxy-2,4-dimethyl-2,4-octadienoic acid (4, 6.3 mg, tR = 12.9 min), (2E,4E)-7-hydroxy-2,4,7-trimethyl-2,4-octadienoic acid (5, 8.0 mg, tR = 13.5 min), and (2E,4E)-2,4-dimethyl-2,4-octadienoic acid (1, 5.2 mg, tR = 30.0 min), respectively. The fifth fraction (424 mg) eluted by CHCl3/MeOH (2:1) from the silica gel column was similarly fractionated by ODS column chromatography. The sixth fraction was purified by the HPLC operation to give (2E,4E)-2,4,7-trimethyl-2,4-octadienoic acid (2, 2.3 mg, tR = 29.1 min) and (R)-(−)-phialomustin B (3, 1.0 mg, tR = 32.1 min).
(2E,4E)-2,4-Dimethyl-2,4-octadienoic acid (1): yellow oil; UV (MeOH) λmax (log ε) 264 (4.08) nm; IR (ATR) νmax: 2959, 2610, 1679, 1622, 1417, 1269, 1138, 1012, 926 cm–1; 1H and 13C NMR data, see Table 1 and Table 2; HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C10H16NaO2, 191.1043; found, 191.1044.
(2E,4E)-2,4,7-Trimethyl-2,4-octadienoic acid (2): yellow oil; UV (MeOH) λmax (log ε) 260 (4.04) nm; IR (ATR) νmax: 2957, 1681, 1622, 1417, 1269, 1137 cm–1; 1H and 13C NMR data, see Table 1 and Table 2; HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C11H18NaO2, 205.1199; found, 205.1202.
(R)-(−)-Phialomustin B (3): yellow oil; [α]D27 −12 (c 0.035, CHCl3), [α]D22 −76 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 256 (4.19) nm; IR (ATR) νmax: 2958, 2956, 1686, 1620, 1279, 982 cm–1; 1H and 13C NMR data, see Table 1 and Table 2; HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C12H20NaO2, 219.1356; found, 219.1356.
(2E,4E)-7-Hydroxy-2,4-dimethyl-2,4-octadienoic acid (4), R/S = 44:56: yellow oil; [α]D22 −2.6 (c 0.30, MeOH); UV (MeOH) λmax (log ε) 259 (4.20) nm; IR (ATR) νmax: 3356, 2969, 2929, 1680, 1623, 1266, 1125 cm–1; 1H and 13C NMR data, see Table 1 and Table 2; HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C10H16NaO3, 207.0992; found, 207.0992.
(2E,4E)-7-Hydroxy-2,4,7-trimethyl-2,4-octadienoic acid (5): yellow oil; UV (MeOH) λmax (log ε) 258 (3.94) nm; IR (ATR) νmax: 3365, 2970, 1681, 1623, 1266, 1130 cm–1; 1H and 13C NMR data, see Table 1 and Table 2; HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C11H18NaO3, 221.1148; found, 221.1149.
Isolation of 6
Compound 6 was obtained from a culture fermented in modified V22 production medium with a composition of soluble starch 1%, glucose 0.5%, NZ Amine, Type A 0.3%, yeast extract 0.2%, Tryptone 0.5% K2HPO4 0.1%, MgSO4·7H2O 0.05%, and CaCO3 0.3%. The pH of the medium was adjusted to 7.0 before autoclaving. At the end of fermentation, 100 mL of 1-butanol was added to each flask, and the flasks were allowed to shake for 1 h. The mixture was centrifuged at 6,000 rpm for 10 min and the organic layer was separated from the aqueous layer containing the mycelium. The organic layer was concentrated in vacuo to give 5.2 g of an extract from a 6 L culture. The crude extract was chromatographed on a silica gel column similarly as above and the sixth fraction was fractionated by ODS column chromatography with a gradient of MeCN/0.1% HCO2H solution (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2 v/v). The fraction 4 (5:5) was evaporated, and purified by preparative HPLC operated at the same conditions as above to give 6-(3,3-dimethylallyl)-N-acetyl-ʟ-tryptophan (6, 45 mg, tR = 10.4 min).
6-(3,3-Dimethylallyl)-N-acetyl-ʟ-tryptophan (6): yellow oil; [α]D22 +15 (c 1.0, MeOH); UV (MeOH) λmax (log ε) 229 (4.59), 282 (4.05) nm; IR (ATR) νmax: 3310, 2916, 1724, 1656, 1627, 1548, 1453, 1223, 806 cm–1; 1H and 13C NMR data, see Table 3; HR–ESI–TOFMS (m/z): [M − H]− calcd for C18H21N2O3, 313.1558; found, 313.1556.
Preparation of methyl ester 4'
To a solution of 4 (0.5 mg, 2.7 μmol) in CHCl3/MeOH (25 μL each) was added a solution of TMS-diazomethane in n-hexane (2.0 M, 25 μL) at room temperature. After stirring for 15 min, the reaction mixture was concentrated to dryness to give methyl ester 4' (0.2 mg).
Methyl ester 4': 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H, -CO2CH3), 7.14 (brs 1H, H3), 5.66 (t, J = 7.4 Hz, 1H, H5), 2.33 (m, 2H, H6), 3.92 (m, 1H, H7), 1.24 (d, J = 6.4 Hz, 3H, H8), 2.01 (d, J = 2.0 Hz, 3H, H9), 1.87 (s, 3H, H10).
Preparation of (R)- and (S)-MPA esters 4'a–d
To a solution of 4' (0.2 mg, 1.0 μmol), (R)-MPA (2.0 mg, 12 μmol), and N,N-dimethyl-4-aminopyridine (3.9 mg, 32 μmol) in dichloromethane (200 μL) were added N,N’-diisopropylcarbodiimide (3 μL). After stirring for 1 h at room temperature, ice-water was poured into the reaction mixture, which was then extracted with EtOAc. After evaporation of the solvent, the residue was purified by preparative silica gel thin-layer chromatography (Kieselgel 60F254; Merck Co.) developed by a mixture of CHCl3/MeOH (5:1) to give (R)-MPA esters 4'a and 4'b with a slight excess yield of the former. A recursive application of Trost’s chiral anisotropy analysis allowed to identify 4'a to be (R)-MPA ester of S- and 4'b to be that of R-enantiomers, respectively.
(R)-MPA ester of S-enantiomer 4'a: 1H NMR (500 MHz, CDCl3) δ 3.77 (s, 3H, -CO2CH3), 7.07 (brs 3H, H3), 5.49 (t, J = 7.7 Hz, 1H, H5), 2.37 (m, 1H, H6a), 2.45 (m, 1H, H6b), 5.03 (m, 1H, H7), 1.14 (d, J = 6.3 Hz, 3H, H8), 1.95 (d, J = 1.3 Hz, 3H, H9), 1.81 (s, 3H, H10), 3.405 (s, 3H, MPA-OCH3), 4.72 (s, 1H, MPA-CαH).
(R)-MPA ester of R-enantiomer 4'b: 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H, -CO2CH3), 6.93 (brs 3H, H3), 5.26 (t, J = 7.7 Hz, 1H, H5), 2.27 (m, 1H, H6a), 2.31 (m, 1H, H6b), 5.03 (m, 1H, H7), 1.26 (d, J = 6.3 Hz, 3H, H8), 1.88 (d, J = 1.3 Hz, 3H, H9), 1.68 (s, 3H, H10), 3.396 (s, 3H, MPA-OCH3), 4.71 (s, 1H, MPA-CαH).
In the same manner as described for the preparation of 4'a and 4'b, a diastereomeric mixture of (S)-MPA esters 4'c and 4'd was prepared from 4' and (S)-MPA.
(S)-MPA ester of 7S enantiomer 4'c: 1H NMR data was identical to those of 4'b.
(S)-MPA ester of 7R enantiomer 4'd: 1H NMR data was identical to those of 4'a.
Chiral HPLC analysis
A 0.2 μL-portion of 4', dissolved in iPrOH, was injected into a cellulose tribenzoate-coated silica gel column (CHIRALCEL OB-H, 4.6 mm × 250 mm, Daicel Co.) eluted with n-hexane/iPrOH (4:1) at 0.5 mL/min. Peaks for (S)- and (R)-4' were detected at 11.3 min and 14.3 min, respectively, by monitoring the absorbance at 250 nm.
Preparation of PGME amides 6a and 6b
In a manner similar to a procedure from [21], to a solution of 6 (1.0 mg, 3.2 μmol) in dry N,N-dimethylformamide (100 μL) and N,N-diisopropylethylamine (10 μL) were added (R)-PGME (1.9 mg, 9.6 μmol), PyBOP (3.6 mg, 7.0 μmol) and HOBt (1.0 mg, 7.4 μmol) at room temperature. After stirring for 3 h, ice-water was poured into the reaction mixture, which was then extracted with EtOAc. After evaporation of the solvent, the residue was purified on a silica gel thin-layer plate (Kieselgel 60F254; Merck Co.) developed by a mixture of CHCl3/MeOH (5:1). Extraction of the collected silica gel powder with MeOH gave (R)-PGME amide 6a (0.5 mg).
(R)-PGME amide 6a: 1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H, NH-1), 7.01 (s, 1H, H-2), 7.46 (d, J = 8.1 Hz, 1H, H-4), 6.78 (d, J = 7.9 Hz, 1H, H-5), 2.85 (dd, J = 14.3, 8.4 Hz, 1H, H-8a), 2.97 (dd, J = 14.0, 5.1 Hz, 1H, H-8b), 4.72 (m, 1H, H-9), 8.02 (d, J = 8.5 Hz, NH-11), 1.79 (s, 3H, H-13); HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C27H31N3NaO4, 484.2207; found, 484.2209.
In the same manner as described for 6a, 6b (0.4 mg) was prepared from 1 and (S)-PGME.
(S)-PGME amide 6b: 1H NMR (500 MHz, DMSO-d6) δ 10.64 (s, 1H, NH-1), 7.07 (d, J = 2.5 Hz, 1H, H-2), 7.53 (d, J = 8.0 Hz, 1H, H-4), 6.80 (d, J = 8.1 Hz, 1H, H-5), 2.88 (dd, J = 14.8, 9.5 Hz, 1H, H-8a), 3.06 (dd, J = 14.8, 4.8 Hz, 1H, H-8b), 4.68 (m, 1H, H-9), 8.01 (d, J = 8.3 Hz, 1H, NH-11), 1.73 (s, 3H, H-13); HR–ESI–TOFMS (m/z): [M + Na]+ calcd for C27H31N3NaO4, 484.2207; found, 484.2207.
Cytotoxicity assay
The cytotoxicity assay was carried out against P388 murine leukemia cells in the same manner as reported previously [36]. The IC50 of a reference drug doxorubicin hydrochloride was 0.13 μM.
Antimicrobial assay
Antimicrobial assays were carried out against five bacteria, K. rhizophila ATCC 9341, S. aureus FDA209P JC-1, E. coli NIHJ JC-2, R. solanacearum SUPP1541, and R. radiobacter NBRC 14554, and a yeast C. albicans NBRC 0197 in the same manner as reported previously [36]. The MIC of the reference antibiotic kanamycin was 0.31 μg/mL (against K. rhizophila).
Supporting Information
| Supporting Information File 1: Copies of NMR spectra. | ||
| Format: PDF | Size: 3.3 MB | Download |
References
-
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2016, 79, 629–661. doi:10.1021/acs.jnatprod.5b01055
Return to citation in text: [1] -
Gonzalez Ronquillo, M.; Angeles Hernandez, J. C. Food Control 2017, 72B, 255–267. doi:10.1016/j.foodcont.2016.03.001
Return to citation in text: [1] -
Demain, A. L.; Sanchez, S. J. Antibiot. 2009, 62, 5–16. doi:10.1038/ja.2008.16
Return to citation in text: [1] -
Villalobos-Delgado, L. H.; Nevárez-Moorillon, G. V.; Caro, I.; Quinto, E. J.; Mateo, J. Natural antimicrobial agents to improve foods shelf life. In Food Quality and Shelf Life; Galanakis, C. M., Ed.; Elsevier: Amsterdam, Netherlands, 2019; pp 125–157. doi:10.1016/b978-0-12-817190-5.00004-5
Return to citation in text: [1] -
Srianta, I.; Ristiarini, S.; Nugerahani, I.; Sen, S. K.; Zhang, B. B.; Xu, G. R.; Blanc, P. J. Int. Food Res. J. 2014, 21, 1–12.
Return to citation in text: [1] -
Zimbro, M. J.; Power, D. A.; Miller, S. M.; Wilson, G. E.; Johnson, J. A. Difco & BBL Manual. Manual of Microbiological Culture Media, 2nd ed.; BD Diagnostics – Diagnostic Systems: Sparks, MD, USA, 2009.
Return to citation in text: [1] -
Ryu, A. H.; Eckalbar, W. L.; Kreimer, A.; Yosef, N.; Ahituv, N. Sci. Rep. 2017, 7, No. 7533. doi:10.1038/s41598-017-07757-w
Return to citation in text: [1] -
Cell Culture Antibiotic Selection Guide. https://www.sigmaaldrich.com/life-science/cell-culture/learning-center/antibiotic-selector.html (accessed Oct 10, 2021).
Return to citation in text: [1] -
Purich, D. L. The Inhibitor Index: A Desk Reference on Enzyme Inhibitors, Receptor Antagonists, Drugs, Toxins, Poisons, Biologics, and Therapeutic Leads, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017. doi:10.1201/9781315184289
Return to citation in text: [1] -
Tiwari, K.; Gupta, R. K. Crit. Rev. Biotechnol. 2012, 32, 108–132. doi:10.3109/07388551.2011.562482
Return to citation in text: [1] -
Dictionary of Natural Products 29.2 Chemical Search. https://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml (accessed Oct 6, 2021).
Return to citation in text: [1] [2] [3] -
Hayakawa, M.; Ishizawa, K.; Nonomura, H. J. Ferment. Technol. 1988, 66, 367–373. doi:10.1016/0385-6380(88)90001-5
Return to citation in text: [1] -
Chen, C.; Chen, Y.; Wu, P.; Chen, B. J. Formosan Med. Assoc. 2014, 113, 72–82. doi:10.1016/j.jfma.2013.10.002
Return to citation in text: [1] -
Laube, H. Clin. Drug Invest. 2002, 22, 141–156. doi:10.2165/00044011-200222030-00001
Return to citation in text: [1] -
Butler, M. S.; Hansford, K. A.; Blaskovich, M. A. T.; Halai, R.; Cooper, M. A. J. Antibiot. 2014, 67, 631–644. doi:10.1038/ja.2014.111
Return to citation in text: [1] -
Shor, B.; Gerber, H.-P.; Sapra, P. Mol. Immunol. 2015, 67, 107–116. doi:10.1016/j.molimm.2014.09.014
Return to citation in text: [1] -
Duggan, S. T. Drugs 2011, 71, 2445–2456. doi:10.2165/11208220-000000000-00000
Return to citation in text: [1] -
List of prokaryotic names with standing in nomenclature. http://www.bacterio.net (accessed Oct 6, 2021).
Return to citation in text: [1] -
Choi, S.-S.; Kim, H.-J.; Lee, H.-S.; Kim, P.; Kim, E.-S. Process Biochem. (Oxford, U. K.) 2015, 50, 1184–1193. doi:10.1016/j.procbio.2015.04.008
Return to citation in text: [1] -
NBRC web site. https://www.nite.go.jp/nbrc/ (accessed March 22, 2021).
Return to citation in text: [1] [2] -
Saito, S.; Atsumi, K.; Zhou, T.; Fukaya, K.; Urabe, D.; Oku, N.; Karim, M. R. U.; Komaki, H.; Igarashi, Y. Beilstein J. Org. Chem. 2020, 16, 1100–1110. doi:10.3762/bjoc.16.97
Return to citation in text: [1] [2] -
Shun, S.; Oku, N.; Igarashi, Y. J. Antibiot. 2022, 75, 44–47.
Return to citation in text: [1] -
Tamura, T.; Nakagaito, Y.; Nishii, T.; Hasegawa, T.; Stackebrandt, E.; Yokota, A. Int. J. Syst. Bacteriol. 1994, 44, 193–203. doi:10.1099/00207713-44-2-193
Return to citation in text: [1] -
Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S. Y.; Medema, M. H.; Weber, T. Nucleic Acids Res. 2019, 47, W81–W87. doi:10.1093/nar/gkz310
Return to citation in text: [1] -
Sheehan, J.; Murphy, C. D.; Caffrey, P. Mol. BioSyst. 2017, 13, 866–873. doi:10.1039/c7mb00112f
Return to citation in text: [1] -
Nalli, Y.; Mirza, D. N.; Wani, Z. A.; Wadhwa, B.; Mallik, F. A.; Raina, C.; chaubey, A.; Riyaz-Ul-Hassan, S.; Ali, A. RSC Adv. 2015, 5, 95307–95312. doi:10.1039/c5ra18121f
Return to citation in text: [1] [2] [3] -
Yabuuchi, T.; Kusumi, T. J. Org. Chem. 2000, 65, 397–404. doi:10.1021/jo991218a
Return to citation in text: [1] -
Kaiser, D.; Onken, U.; Sattler, I.; Zeeck, A. Appl. Microbiol. Biotechnol. 1994, 41, 309–312. doi:10.1007/bf00221224
Return to citation in text: [1] -
Sattler, I.; Gröne, C.; Zeeck, A. J. Org. Chem. 1993, 58, 6583–6587. doi:10.1021/jo00076a015
Return to citation in text: [1] -
Kohno, J.; Nishio, M.; Kawano, K.; Nakanishi, N.; Suzuki, S.; Uchida, T.; Komatsubara, S. J. Antibiot. 1996, 49, 1212–1220. doi:10.7164/antibiotics.49.1212
Return to citation in text: [1] -
Guo, X. W.; Zhang, J.; Li, J. S. Nat. Prod. Res. Dev. 2016, 28, 481–485.
Return to citation in text: [1] -
Awakawa, T.; Crüsemann, M.; Munguia, J.; Ziemert, N.; Nizet, V.; Fenical, W.; Moore, B. S. ChemBioChem 2015, 16, 1443–1447. doi:10.1002/cbic.201500177
Return to citation in text: [1] -
Wang, B.; Guo, F.; Huang, C.; Zhao, H. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 8449–8454. doi:10.1073/pnas.1917664117
Return to citation in text: [1] -
Fan, A.; Winkelblech, J.; Li, S.-M. Appl. Microbiol. Biotechnol. 2015, 99, 7399–7415. doi:10.1007/s00253-015-6813-9
Return to citation in text: [1] -
Takahashi, S.; Takagi, H.; Toyoda, A.; Uramoto, M.; Nogawa, T.; Ueki, M.; Sakaki, Y.; Osada, H. J. Bacteriol. 2010, 192, 2839–2851. doi:10.1128/jb.01557-09
Return to citation in text: [1] -
Karim, M. R. U.; Harunari, E.; Oku, N.; Akasaka, K.; Igarashi, Y. J. Nat. Prod. 2020, 83, 1295–1299. doi:10.1021/acs.jnatprod.0c00082
Return to citation in text: [1] [2]
| 29. | Sattler, I.; Gröne, C.; Zeeck, A. J. Org. Chem. 1993, 58, 6583–6587. doi:10.1021/jo00076a015 |
| 30. | Kohno, J.; Nishio, M.; Kawano, K.; Nakanishi, N.; Suzuki, S.; Uchida, T.; Komatsubara, S. J. Antibiot. 1996, 49, 1212–1220. doi:10.7164/antibiotics.49.1212 |
| 26. | Nalli, Y.; Mirza, D. N.; Wani, Z. A.; Wadhwa, B.; Mallik, F. A.; Raina, C.; chaubey, A.; Riyaz-Ul-Hassan, S.; Ali, A. RSC Adv. 2015, 5, 95307–95312. doi:10.1039/c5ra18121f |
| 1. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2016, 79, 629–661. doi:10.1021/acs.jnatprod.5b01055 |
| 2. | Gonzalez Ronquillo, M.; Angeles Hernandez, J. C. Food Control 2017, 72B, 255–267. doi:10.1016/j.foodcont.2016.03.001 |
| 10. | Tiwari, K.; Gupta, R. K. Crit. Rev. Biotechnol. 2012, 32, 108–132. doi:10.3109/07388551.2011.562482 |
| 11. | Dictionary of Natural Products 29.2 Chemical Search. https://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml (accessed Oct 6, 2021). |
| 36. | Karim, M. R. U.; Harunari, E.; Oku, N.; Akasaka, K.; Igarashi, Y. J. Nat. Prod. 2020, 83, 1295–1299. doi:10.1021/acs.jnatprod.0c00082 |
| 6. | Zimbro, M. J.; Power, D. A.; Miller, S. M.; Wilson, G. E.; Johnson, J. A. Difco & BBL Manual. Manual of Microbiological Culture Media, 2nd ed.; BD Diagnostics – Diagnostic Systems: Sparks, MD, USA, 2009. |
| 7. | Ryu, A. H.; Eckalbar, W. L.; Kreimer, A.; Yosef, N.; Ahituv, N. Sci. Rep. 2017, 7, No. 7533. doi:10.1038/s41598-017-07757-w |
| 8. | Cell Culture Antibiotic Selection Guide. https://www.sigmaaldrich.com/life-science/cell-culture/learning-center/antibiotic-selector.html (accessed Oct 10, 2021). |
| 9. | Purich, D. L. The Inhibitor Index: A Desk Reference on Enzyme Inhibitors, Receptor Antagonists, Drugs, Toxins, Poisons, Biologics, and Therapeutic Leads, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017. doi:10.1201/9781315184289 |
| 19. | Choi, S.-S.; Kim, H.-J.; Lee, H.-S.; Kim, P.; Kim, E.-S. Process Biochem. (Oxford, U. K.) 2015, 50, 1184–1193. doi:10.1016/j.procbio.2015.04.008 |
| 36. | Karim, M. R. U.; Harunari, E.; Oku, N.; Akasaka, K.; Igarashi, Y. J. Nat. Prod. 2020, 83, 1295–1299. doi:10.1021/acs.jnatprod.0c00082 |
| 4. | Villalobos-Delgado, L. H.; Nevárez-Moorillon, G. V.; Caro, I.; Quinto, E. J.; Mateo, J. Natural antimicrobial agents to improve foods shelf life. In Food Quality and Shelf Life; Galanakis, C. M., Ed.; Elsevier: Amsterdam, Netherlands, 2019; pp 125–157. doi:10.1016/b978-0-12-817190-5.00004-5 |
| 5. | Srianta, I.; Ristiarini, S.; Nugerahani, I.; Sen, S. K.; Zhang, B. B.; Xu, G. R.; Blanc, P. J. Int. Food Res. J. 2014, 21, 1–12. |
| 17. | Duggan, S. T. Drugs 2011, 71, 2445–2456. doi:10.2165/11208220-000000000-00000 |
| 35. | Takahashi, S.; Takagi, H.; Toyoda, A.; Uramoto, M.; Nogawa, T.; Ueki, M.; Sakaki, Y.; Osada, H. J. Bacteriol. 2010, 192, 2839–2851. doi:10.1128/jb.01557-09 |
| 18. | List of prokaryotic names with standing in nomenclature. http://www.bacterio.net (accessed Oct 6, 2021). |
| 21. | Saito, S.; Atsumi, K.; Zhou, T.; Fukaya, K.; Urabe, D.; Oku, N.; Karim, M. R. U.; Komaki, H.; Igarashi, Y. Beilstein J. Org. Chem. 2020, 16, 1100–1110. doi:10.3762/bjoc.16.97 |
| 13. | Chen, C.; Chen, Y.; Wu, P.; Chen, B. J. Formosan Med. Assoc. 2014, 113, 72–82. doi:10.1016/j.jfma.2013.10.002 |
| 15. | Butler, M. S.; Hansford, K. A.; Blaskovich, M. A. T.; Halai, R.; Cooper, M. A. J. Antibiot. 2014, 67, 631–644. doi:10.1038/ja.2014.111 |
| 33. | Wang, B.; Guo, F.; Huang, C.; Zhao, H. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 8449–8454. doi:10.1073/pnas.1917664117 |
| 11. | Dictionary of Natural Products 29.2 Chemical Search. https://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml (accessed Oct 6, 2021). |
| 16. | Shor, B.; Gerber, H.-P.; Sapra, P. Mol. Immunol. 2015, 67, 107–116. doi:10.1016/j.molimm.2014.09.014 |
| 34. | Fan, A.; Winkelblech, J.; Li, S.-M. Appl. Microbiol. Biotechnol. 2015, 99, 7399–7415. doi:10.1007/s00253-015-6813-9 |
| 12. | Hayakawa, M.; Ishizawa, K.; Nonomura, H. J. Ferment. Technol. 1988, 66, 367–373. doi:10.1016/0385-6380(88)90001-5 |
| 11. | Dictionary of Natural Products 29.2 Chemical Search. https://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml (accessed Oct 6, 2021). |
| 14. | Laube, H. Clin. Drug Invest. 2002, 22, 141–156. doi:10.2165/00044011-200222030-00001 |
| 32. | Awakawa, T.; Crüsemann, M.; Munguia, J.; Ziemert, N.; Nizet, V.; Fenical, W.; Moore, B. S. ChemBioChem 2015, 16, 1443–1447. doi:10.1002/cbic.201500177 |
| 21. | Saito, S.; Atsumi, K.; Zhou, T.; Fukaya, K.; Urabe, D.; Oku, N.; Karim, M. R. U.; Komaki, H.; Igarashi, Y. Beilstein J. Org. Chem. 2020, 16, 1100–1110. doi:10.3762/bjoc.16.97 |
| 27. | Yabuuchi, T.; Kusumi, T. J. Org. Chem. 2000, 65, 397–404. doi:10.1021/jo991218a |
| 28. | Kaiser, D.; Onken, U.; Sattler, I.; Zeeck, A. Appl. Microbiol. Biotechnol. 1994, 41, 309–312. doi:10.1007/bf00221224 |
| 26. | Nalli, Y.; Mirza, D. N.; Wani, Z. A.; Wadhwa, B.; Mallik, F. A.; Raina, C.; chaubey, A.; Riyaz-Ul-Hassan, S.; Ali, A. RSC Adv. 2015, 5, 95307–95312. doi:10.1039/c5ra18121f |
| 26. | Nalli, Y.; Mirza, D. N.; Wani, Z. A.; Wadhwa, B.; Mallik, F. A.; Raina, C.; chaubey, A.; Riyaz-Ul-Hassan, S.; Ali, A. RSC Adv. 2015, 5, 95307–95312. doi:10.1039/c5ra18121f |
| 25. | Sheehan, J.; Murphy, C. D.; Caffrey, P. Mol. BioSyst. 2017, 13, 866–873. doi:10.1039/c7mb00112f |
| 23. | Tamura, T.; Nakagaito, Y.; Nishii, T.; Hasegawa, T.; Stackebrandt, E.; Yokota, A. Int. J. Syst. Bacteriol. 1994, 44, 193–203. doi:10.1099/00207713-44-2-193 |
| 24. | Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S. Y.; Medema, M. H.; Weber, T. Nucleic Acids Res. 2019, 47, W81–W87. doi:10.1093/nar/gkz310 |
© 2021 Saito et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.