Abstract
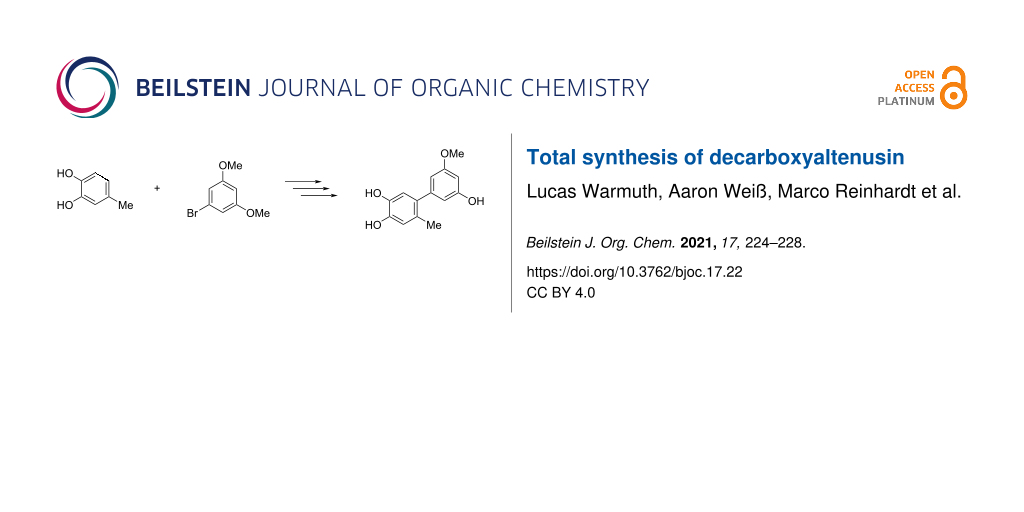
The total synthesis of decarboxyaltenusin (5’-methoxy-6-methyl-[1,1’-biphenyl]-3,3’,4-triol), a toxin produced by various mold fungi, has been achieved in seven steps in a yield of 31% starting from 4-methylcatechol and 1-bromo-3,5-dimethoxybenzene, where the longest linear sequence consists of five steps. The key reaction was a palladium-catalyzed Suzuki coupling of an aromatic boronate with a brominated resorcin derivative.

Graphical Abstract
Introduction
5’-Methoxy-6-methyl-[1,1’-biphenyl]-3,3’,4-triol (1, Scheme 1) has been first mentioned 1974 as a reduction and decarboxylation product of dehydroaltenusin [1]. As the compound has later been isolated from Ulocladium sp. [2], Nigrospora sphaerica, Phialophora sp. [3], Penicillium pinophilum SD-272 [4], Alternaria sp. [3,5,6], and from the endophytic fungus Botryosphaeria dothidea in Melia azedarach [7] it turned out to be a widely occurring natural product. It shows marked DPPH radical scavenging activities with determined IC50 values of 18.7 ± 0.2 μM [7] and 148 ± 3 μM [2], respectively, and displays inhibitory activity against three tyrosine kinases (EGFR, VEGFR-1, and c-Met) [5]. As mentioned, it is accessible through the reduction of dehydroaltenusin with zinc powder in acetic acid with intermediate formation of altenusin [1]. Nevertheless, this route cannot be considered as a viable approach to this compound due to the reduced accessibility of the precursors and since no experimental details have been published for the transformation. To continue our efforts in the total synthesis of mycotoxins [8-18] and to provide larger amounts of the polyketide 1 sufficient for thorough biological investigations (as have been suggested by the European Food Safety Authority, EFSA [19]), we considered it useful to supply a more straightforward synthesis of this compound, for which we here propose the obvious name decarboxyaltenusin.
![[1860-5397-17-22-i1]](/bjoc/content/inline/1860-5397-17-22-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
In a retrosynthetic analysis, we envisioned a Suzuki coupling of two suitably substituted arenes. Silyl protecting groups like the tert-butyldimethylsilyl group (TBS) were considered appropriate for all projected reaction steps. The boronate moiety 6a was prepared starting with 4-methylcatechol (2), which was initially protected with tert-butyldimethylsilyl chloride in the presence of 4-(dimethylamino)pyridine (DMAP) and imidazole (Scheme 2) according to a published procedure [20]. The thus obtained bis(silylether) 3 was then brominated with N-bromosuccinimide (NBS), where the utilization of acetonitrile as solvent [21] instead of carbon tetrachloride [22] furnished a close to quantitative yield of bromide 5a, though a prolonged reaction time of 72 h had to be accepted. The subsequent formation of boronate 6a was accomplished through a metal–halogen interchange with butyllithium and trapping with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane [23].
![[1860-5397-17-22-i2]](/bjoc/content/inline/1860-5397-17-22-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of arylboronates 6. Conditions: a) TBSCl, DMAP, imidazole, DMF, 50 °C, 4 h (96%); b) NBS, MeCN, rt, 71 h (quant.); c) NBS, MeCN, rt, 72 h (R = TBS, 96%); d) BnBr, KI, K2CO3, DMF/acetone, 70 °C, 29 h, (R = Bn, 86%); e) R = TBS: BuLi, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF, −78 °C, 0.45–2 h, rt, 18 h (57%); R = Bn: bis(pinacolato)diboron, Pd(dppf)Cl2·CH2Cl2 (cat.), KOAc, dioxane, 80 °C, 17 h (55%).
Scheme 2: Synthesis of arylboronates 6. Conditions: a) TBSCl, DMAP, imidazole, DMF, 50 °C, 4 h (96%); b) NBS,...
The electrophilic compound suitable for the projected cross coupling was obtained by mono-demethylation of commercially available 1-bromo-3,5-dimethoxybenzene (7) with boron tribromide (Scheme 3). A satisfactory yield of phenol 8 was observed with 0.9 equivalents of the Lewis acid, while the utilization of 1.5 equivalents led to a significant overreaction with the predominant formation of 5-bromobenzene-1,3-diol (59%) together with a smaller amount of the required product 8 (30%). The subsequent protection with the TBS group yielded the known aryl bromide 9a [24] with a 73% yield.
![[1860-5397-17-22-i3]](/bjoc/content/inline/1860-5397-17-22-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of aryl bromides 9. Conditions: f) BBr3, −78 °C to rt, 18 h (71%); g) R = TBS: TBSCl, DMAP, imidazole, DMF, 55 °C, 4 h (73%); R = Bn: BnBr, K2CO3, DMF/acetone 1:2, 80 °C, 43 h (98%).
Scheme 3: Synthesis of aryl bromides 9. Conditions: f) BBr3, −78 °C to rt, 18 h (71%); g) R = TBS: TBSCl, DMA...
The Suzuki coupling of boronate 6a and aryl bromide 9a using palladium acetate and cesium carbonate in the presence of the ligand SPhos [25] yielded biaryl 10a with virtually quantitative yield (98%, Scheme 4). Unfortunately, the removal of non-identified byproducts and of 9a (which had been used in a 1.2-fold excess) turned out to be very laborious. Moreover, the subsequent deprotection to the natural product 1 could not be achieved sufficiently: After treatment of 10a with tetrabutylammonium fluoride (TBAF), the signals of 1 could be detected in a 1H NMR spectrum of the crude product, but purification and isolation of the product by column chromatography was not possible ‒ neither with conventional nor with reversed phase methods. It turned out that the high polarity of the triol complicated its separation from other polar side products.
![[1860-5397-17-22-i4]](/bjoc/content/inline/1860-5397-17-22-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Final steps in the synthesis of biaryl 1. Conditions: h) Pd(OAc)2, SPhos, Cs2CO3, dioxane/H2O 7:1, 70 °C, 18 h, (R = TBS: 98%, containing non-separable impurities; R = Bn: 89%) ; i) R = Bn: Pd/C (10%), H2, THF, 8 bar, 24 h, 40 °C (88%).
Scheme 4: Final steps in the synthesis of biaryl 1. Conditions: h) Pd(OAc)2, SPhos, Cs2CO3, dioxane/H2O 7:1, ...
To circumvent this problem, we decided to use a different protection group strategy and to employ hydrogenolytically cleavable benzyl groups. The synthesis of the benzyl-protected boronate was here achieved with a modified strategy including bromination [21] of 4-methylcatechol (2) to the known bromide 4 [26] and subsequent benzyl protection to the bis(benzyl ether) 5b using standard conditions (Scheme 2) [27]. The preparation of boronate 6b applying the conditions used for the silylated substrate 6a (vide supra) led to a mediocre 44% yield, but the utilization of a palladium-catalyzed borylation with bis(pinacolato)diboron afforded the product with 55% yield. The O-benzylation of phenol 8 furnishing the bromide 9b was accomplished with virtually quantitative yield. Suzuki coupling of the benzyl-protected compounds 6b and 9b led to biaryl 10b with 89% yield; it was deprotected with palladium on charcoal under eight bar hydrogen pressure (Scheme 4). The product 1 could now be purified by a simple chromatography on silica gel and was obtained in 88% yield.
NMR spectroscopic data of the natural product had been published by Wang et al. [2] and by Xiao et al. [7] (Table 1). A comparison of the 13C NMR data of the now synthesized compound with Wang’s data shows a systematic deviation of about −0.5 ppm (possibly due to a calibration inaccuracy in the original paper) and a deviant signal around 144 ppm, which is a further −0.4 ppm off (marked in boldface). Nevertheless, all signals published by Xiao et al. are in virtually perfect agreement with the NMR data measured by us, what leaves no reasonable doubt that the synthesized structure 1 is identical with the natural product.
Table 1: NMR data of natural and synthetic biaryl 1.
| natural product | synthetic product | deviationa | ||
| δ [ppm]b,c | δ [ppm]c,d | signal | δ [ppm] | Δδ [ppm] |
| 19.8 | 19.50 | 6-Me | 19.4 | −0.4/−0.1 |
| 55.4 | 54.97 | OMe | 54.9 | −0.5/−0.1 |
| 99.7 | 99.21 | C-4’ | 99.2 | −0.5/±0.0 |
| 106.2 | 105.80 | C-2’ | 105.8 | −0.4/±0.0 |
| 109.3 | 108.84 | C-6’ | 108.8 | −0.5/±0.0 |
| 117.1 | 116.70 | C-2 | 116.7 | −0.4/±0.0 |
| 118.0 | 117.56 | C-5 | 117.5 | −0.5/−0.1 |
| 125.3 | 124.89 | C-6 | 124.8 | −0.5/−0.1 |
| 132.7 | 132.30 | C-1 | 132.3 | −0.4/±0.0 |
| 143.5e,f | 142.92 | C-1’ | 142.9 | −0.6/±0.0 |
| 144.5e,f | 143.66 | C-3 | 143.6 | −0.9/−0.1 |
| 144.8e,f | 144.39 | C-4 | 144.3 | −0.5/−0.1 |
| 158.5 | 158.06 | C-3’ | 158.0 | −0.5/−0.1 |
| 160.4 | 160.04 | C-5’ | 160.0 | −0.4/±0.0 |
| 2.05 (s) | 6-CH3 | 2.05 (s) | ±0.00 | |
| 3.69 (s) | 5’-OCH3 | 3.70 (s) | +0.01 | |
| 6.20 (br. s) | 6’-H | 6.20 (dd) | ±0.00 | |
| 6.21 (br. s) | 2’-H | 6.22 (t) | +0.01 | |
| 6.25 (br. s) | 4’-H | 6.25 (t) | ±0.00 | |
| 6.54 (s) | 2-H | 6.55 (s) | +0.01 | |
| 6.60 (s) | 5-H | 6.60 (s) | ±0.00 | |
| 8.72 (br. s) | 3-OH | 8.72 (br. s) | ±0.00 | |
| 8.78 (br. s) | 4-OH | 8.78 (br. s) | ±0.00 | |
| 9.42 (br. s) | 3’-OH | 9.37 (br. s) | − 0.05 | |
aDeviation of the synthesized product’s data from published data: Wang et al. [2]/Xiao et al. [7]; bdata published by Wang et al. [2]; cthe data are given in ascending order. Assignments in the original papers are in agreement with those given for the synthesized product (except footnote e); ddata published by Xiao et al. [7]; ethe assignment in the original paper is: 143.5: C-4, 144.5: C-3, and 144.8: C-1’; fa superscript letter is included after these numbers in the original paper, but a corresponding footnote is missing. It can be assumed that the assignment of these signals had been considered questionable.
Decarboxyaltenusin (1) was screened for toxicity towards human HeLa cells but proved nontoxic at biologically relevant concentrations and showed an LD50 value of above 50 μM. This screening was performed by measuring the cell viability using an MTT assay, where the viability is assessed based upon the reduction of the yellow tetrazolium MTT [3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide] by metabolically active and hence viable cells. The resulting intracellular purple formazan was quantified spectrophotometrically. The cell viability was calculated based on an untreated control. The cell viability cut-off was <70%. At a 0.5 μM concentration of decarboxyaltenusin (1), it was 103% ± 1%, at 5 μM it was 94% ± 1.4%, and at 50 μM 92% ± 1%.
Conclusion
The total synthesis of decarboxyaltenusin (1) was achieved in seven steps in a yield of 31% starting from 4-methylcatechol (2) and 1-bromo-3,5-dimethoxybenzene (7), where the longest linear sequence has consisted of five steps. Decarboxyaltenusin turned out to be nontoxic towards human HeLa cells.
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR spectra of all new compounds and of decarboxyaltenusin (1). | ||
| Format: PDF | Size: 2.0 MB | Download |
References
-
Kameda, K.; Aoki, H.; Namiki, M.; Overeem, J. C. Tetrahedron Lett. 1974, 15, 103–106. doi:10.1016/s0040-4039(01)82147-x
Return to citation in text: [1] [2] -
Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013
Return to citation in text: [1] [2] [3] [4] [5] -
He, J.-W.; Chen, G.-D.; Gao, H.; Yang, F.; Li, X.-X.; Peng, T.; Guo, L.-D.; Yao, X.-S. Fitoterapia 2012, 83, 1087–1091. doi:10.1016/j.fitote.2012.05.002
Return to citation in text: [1] [2] -
Wang, M.; Li, X.; Li, C.; Wang, B. Haiyang Kexue 2014, 38, 1–5. doi:10.11759/hykx20130323002
Chem. Abstr. 2015, 162, 652122.
Return to citation in text: [1] -
Guo, L.-z.; Liu, C.-x.; Xu, B.; Chen, J.-f.; Zou, K.; Guo, Z.-y. Huaxue Yanjiu Yu Yingyong 2014, 26, 306–310.
Chem. Abstr. 2014, 162, 445682.
Return to citation in text: [1] [2] -
Chen, B.; Liu, L.; Zhu, X.; Wang, J.; Long, Y.; Jiang, S.-P.; Xu, A.-G.; Lin, Y.-C. Nat. Prod. Res. 2015, 29, 1212–1216. doi:10.1080/14786419.2014.997232
Return to citation in text: [1] -
Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f
Return to citation in text: [1] [2] [3] [4] [5] -
Koch, K.; Podlech, J.; Pfeiffer, E.; Metzler, M. J. Org. Chem. 2005, 70, 3275–3276. doi:10.1021/jo050075r
Return to citation in text: [1] -
Altemöller, M.; Podlech, J.; Fenske, D. Eur. J. Org. Chem. 2006, 1678–1684. doi:10.1002/ejoc.200500904
Return to citation in text: [1] -
Altemöller, M.; Podlech, J. Eur. J. Org. Chem. 2009, 2275–2282. doi:10.1002/ejoc.200900125
Return to citation in text: [1] -
Altemöller, M.; Podlech, J. J. Nat. Prod. 2009, 72, 1288–1290. doi:10.1021/np900265q
Return to citation in text: [1] -
Cudaj, J.; Podlech, J. Tetrahedron Lett. 2010, 51, 3092–3094. doi:10.1016/j.tetlet.2010.04.024
Return to citation in text: [1] -
Cudaj, J.; Podlech, J. Synlett 2012, 23, 371–374. doi:10.1055/s-0031-1290135
Return to citation in text: [1] -
Geiseler, O.; Podlech, J. Tetrahedron 2012, 68, 7280–7287. doi:10.1016/j.tet.2012.06.104
Return to citation in text: [1] -
Nemecek, G.; Cudaj, J.; Podlech, J. Eur. J. Org. Chem. 2012, 3863–3870. doi:10.1002/ejoc.201200506
Return to citation in text: [1] -
Nemecek, G.; Thomas, R.; Goesmann, H.; Feldmann, C.; Podlech, J. Eur. J. Org. Chem. 2013, 6420–6432. doi:10.1002/ejoc.201300879
Return to citation in text: [1] -
Thomas, R.; Nemecek, G.; Podlech, J. Nat. Prod. Res. 2013, 27, 2053–2054. doi:10.1080/14786419.2013.819509
Return to citation in text: [1] -
Kohler, D.; Podlech, J. Eur. J. Org. Chem. 2019, 1748–1753. doi:10.1002/ejoc.201801801
Return to citation in text: [1] -
EFSA Panel on Contaminants in the Food Chain (CONTAM). EFSA J. 2011, 9, 2407. doi:10.2903/j.efsa.2011.2407
Return to citation in text: [1] -
Berkowitz, D. B.; Smith, M. K. J. Org. Chem. 1995, 60, 1233–1238. doi:10.1021/jo00110a029
Return to citation in text: [1] -
Ladouceur, S.; Fortin, D.; Zysman-Colman, E. Inorg. Chem. 2010, 49, 5625–5641. doi:10.1021/ic100521t
Return to citation in text: [1] [2] -
Aciro, C.; Claridge, T. D. W.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3751–3761. doi:10.1039/b808811j
Return to citation in text: [1] -
Li, Y.; Li, A.-Y.; Li, B.-X.; Huang, J.; Zhao, L.; Wang, B.-Z.; Li, J.-W.; Zhu, X.-H.; Peng, J.; Cao, Y.; Ma, D.-G.; Roncali, J. Org. Lett. 2009, 11, 5318–5321. doi:10.1021/ol9022563
Return to citation in text: [1] -
Wada, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. RSC Adv. 2019, 9, 31654–31658. doi:10.1039/c9ra05532k
Return to citation in text: [1] -
Altemöller, M.; Gehring, T.; Cudaj, J.; Podlech, J.; Goesmann, H.; Feldmann, C.; Rothenberger, A. Eur. J. Org. Chem. 2009, 2130–2140. doi:10.1002/ejoc.200801278
Return to citation in text: [1] -
Alig, L.; Alsenz, J.; Andjelkovic, M.; Bendels, S.; Bénardeau, A.; Bleicher, K.; Bourson, A.; David-Pierson, P.; Guba, W.; Hildbrand, S.; Kube, D.; Lübbers, T.; Mayweg, A. V.; Narquizian, R.; Neidhart, W.; Nettekoven, M.; Plancher, J.-M.; Rocha, C.; Rogers-Evans, M.; Röver, S.; Schneider, G.; Taylor, S.; Waldmeier, P. J. Med. Chem. 2008, 51, 2115–2127. doi:10.1021/jm701487t
Return to citation in text: [1] -
da Silva Pinto, A. C.; Silva, L. F. R.; Cavalcanti, B. C.; Melo, M. R. S.; Chaves, F. C. M.; Lotufo, L. V. C.; de Moraes, M. O.; de Andrade-Neto, V. F.; Tadei, W. P.; Pessoa, C. O.; Ribeiro Vieirac, P. P.; Pohlit, A. M. Eur. J. Med. Chem. 2009, 44, 2731–2735. doi:10.1016/j.ejmech.2008.10.025
Return to citation in text: [1]
| 1. | Kameda, K.; Aoki, H.; Namiki, M.; Overeem, J. C. Tetrahedron Lett. 1974, 15, 103–106. doi:10.1016/s0040-4039(01)82147-x |
| 3. | He, J.-W.; Chen, G.-D.; Gao, H.; Yang, F.; Li, X.-X.; Peng, T.; Guo, L.-D.; Yao, X.-S. Fitoterapia 2012, 83, 1087–1091. doi:10.1016/j.fitote.2012.05.002 |
| 5. |
Guo, L.-z.; Liu, C.-x.; Xu, B.; Chen, J.-f.; Zou, K.; Guo, Z.-y. Huaxue Yanjiu Yu Yingyong 2014, 26, 306–310.
Chem. Abstr. 2014, 162, 445682. |
| 6. | Chen, B.; Liu, L.; Zhu, X.; Wang, J.; Long, Y.; Jiang, S.-P.; Xu, A.-G.; Lin, Y.-C. Nat. Prod. Res. 2015, 29, 1212–1216. doi:10.1080/14786419.2014.997232 |
| 22. | Aciro, C.; Claridge, T. D. W.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3751–3761. doi:10.1039/b808811j |
| 4. |
Wang, M.; Li, X.; Li, C.; Wang, B. Haiyang Kexue 2014, 38, 1–5. doi:10.11759/hykx20130323002
Chem. Abstr. 2015, 162, 652122. |
| 23. | Li, Y.; Li, A.-Y.; Li, B.-X.; Huang, J.; Zhao, L.; Wang, B.-Z.; Li, J.-W.; Zhu, X.-H.; Peng, J.; Cao, Y.; Ma, D.-G.; Roncali, J. Org. Lett. 2009, 11, 5318–5321. doi:10.1021/ol9022563 |
| 3. | He, J.-W.; Chen, G.-D.; Gao, H.; Yang, F.; Li, X.-X.; Peng, T.; Guo, L.-D.; Yao, X.-S. Fitoterapia 2012, 83, 1087–1091. doi:10.1016/j.fitote.2012.05.002 |
| 20. | Berkowitz, D. B.; Smith, M. K. J. Org. Chem. 1995, 60, 1233–1238. doi:10.1021/jo00110a029 |
| 2. | Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013 |
| 21. | Ladouceur, S.; Fortin, D.; Zysman-Colman, E. Inorg. Chem. 2010, 49, 5625–5641. doi:10.1021/ic100521t |
| 5. |
Guo, L.-z.; Liu, C.-x.; Xu, B.; Chen, J.-f.; Zou, K.; Guo, Z.-y. Huaxue Yanjiu Yu Yingyong 2014, 26, 306–310.
Chem. Abstr. 2014, 162, 445682. |
| 8. | Koch, K.; Podlech, J.; Pfeiffer, E.; Metzler, M. J. Org. Chem. 2005, 70, 3275–3276. doi:10.1021/jo050075r |
| 9. | Altemöller, M.; Podlech, J.; Fenske, D. Eur. J. Org. Chem. 2006, 1678–1684. doi:10.1002/ejoc.200500904 |
| 10. | Altemöller, M.; Podlech, J. Eur. J. Org. Chem. 2009, 2275–2282. doi:10.1002/ejoc.200900125 |
| 11. | Altemöller, M.; Podlech, J. J. Nat. Prod. 2009, 72, 1288–1290. doi:10.1021/np900265q |
| 12. | Cudaj, J.; Podlech, J. Tetrahedron Lett. 2010, 51, 3092–3094. doi:10.1016/j.tetlet.2010.04.024 |
| 13. | Cudaj, J.; Podlech, J. Synlett 2012, 23, 371–374. doi:10.1055/s-0031-1290135 |
| 14. | Geiseler, O.; Podlech, J. Tetrahedron 2012, 68, 7280–7287. doi:10.1016/j.tet.2012.06.104 |
| 15. | Nemecek, G.; Cudaj, J.; Podlech, J. Eur. J. Org. Chem. 2012, 3863–3870. doi:10.1002/ejoc.201200506 |
| 16. | Nemecek, G.; Thomas, R.; Goesmann, H.; Feldmann, C.; Podlech, J. Eur. J. Org. Chem. 2013, 6420–6432. doi:10.1002/ejoc.201300879 |
| 17. | Thomas, R.; Nemecek, G.; Podlech, J. Nat. Prod. Res. 2013, 27, 2053–2054. doi:10.1080/14786419.2013.819509 |
| 18. | Kohler, D.; Podlech, J. Eur. J. Org. Chem. 2019, 1748–1753. doi:10.1002/ejoc.201801801 |
| 2. | Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013 |
| 19. | EFSA Panel on Contaminants in the Food Chain (CONTAM). EFSA J. 2011, 9, 2407. doi:10.2903/j.efsa.2011.2407 |
| 7. | Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f |
| 7. | Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f |
| 1. | Kameda, K.; Aoki, H.; Namiki, M.; Overeem, J. C. Tetrahedron Lett. 1974, 15, 103–106. doi:10.1016/s0040-4039(01)82147-x |
| 21. | Ladouceur, S.; Fortin, D.; Zysman-Colman, E. Inorg. Chem. 2010, 49, 5625–5641. doi:10.1021/ic100521t |
| 24. | Wada, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. RSC Adv. 2019, 9, 31654–31658. doi:10.1039/c9ra05532k |
| 25. | Altemöller, M.; Gehring, T.; Cudaj, J.; Podlech, J.; Goesmann, H.; Feldmann, C.; Rothenberger, A. Eur. J. Org. Chem. 2009, 2130–2140. doi:10.1002/ejoc.200801278 |
| 2. | Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013 |
| 7. | Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f |
| 2. | Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013 |
| 7. | Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f |
| 2. | Wang, Q.-X.; Bao, L.; Yang, X.-L.; Guo, H.; Yang, R.-N.; Ren, B.; Zhang, L.-X.; Dai, H.-Q.; Guo, L.-D.; Liu, H.-W. Fitoterapia 2012, 83, 209–214. doi:10.1016/j.fitote.2011.10.013 |
| 7. | Xiao, J.; Zhang, Q.; Gao, Y.-Q.; Tang, J.-J.; Zhang, A.-L.; Gao, J.-M. J. Agric. Food Chem. 2014, 62, 3584–3590. doi:10.1021/jf500054f |
| 26. | Alig, L.; Alsenz, J.; Andjelkovic, M.; Bendels, S.; Bénardeau, A.; Bleicher, K.; Bourson, A.; David-Pierson, P.; Guba, W.; Hildbrand, S.; Kube, D.; Lübbers, T.; Mayweg, A. V.; Narquizian, R.; Neidhart, W.; Nettekoven, M.; Plancher, J.-M.; Rocha, C.; Rogers-Evans, M.; Röver, S.; Schneider, G.; Taylor, S.; Waldmeier, P. J. Med. Chem. 2008, 51, 2115–2127. doi:10.1021/jm701487t |
| 27. | da Silva Pinto, A. C.; Silva, L. F. R.; Cavalcanti, B. C.; Melo, M. R. S.; Chaves, F. C. M.; Lotufo, L. V. C.; de Moraes, M. O.; de Andrade-Neto, V. F.; Tadei, W. P.; Pessoa, C. O.; Ribeiro Vieirac, P. P.; Pohlit, A. M. Eur. J. Med. Chem. 2009, 44, 2731–2735. doi:10.1016/j.ejmech.2008.10.025 |
© 2021 Warmuth et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)