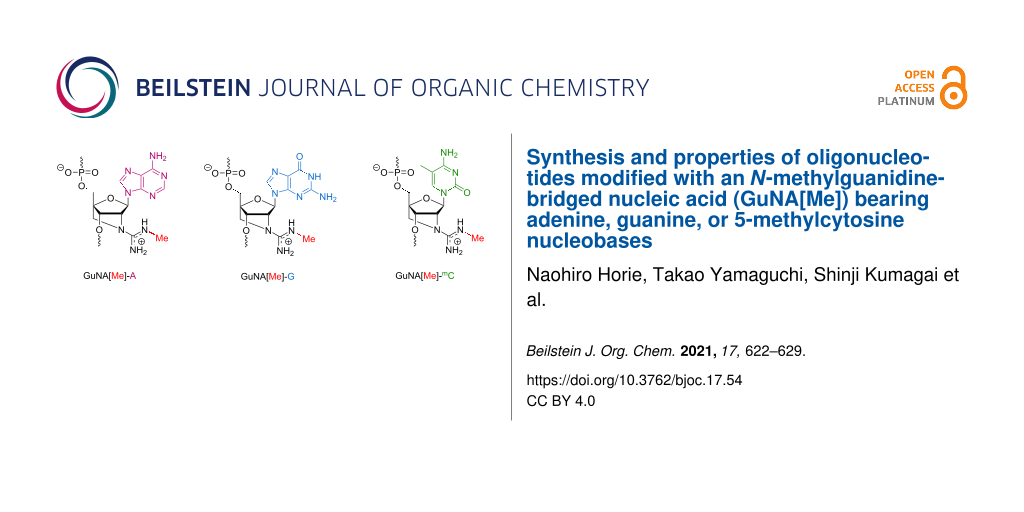
Abstract
Chemical modifications have been extensively used for therapeutic oligonucleotides because they strongly enhance the stability against nucleases, binding affinity to the targets, and efficacy. We previously reported that oligonucleotides modified with an N-methylguanidine-bridged nucleic acid (GuNA[Me]) bearing the thymine (T) nucleobase show excellent biophysical properties for applications in antisense technology. In this paper, we describe the synthesis of GuNA[Me] phosphoramidites bearing other typical nucleobases including adenine (A), guanine (G), and 5-methylcytosine (mC). The phosphoramidites were successfully incorporated into oligonucleotides following the method previously developed for the GuNA[Me]-T-modified oligonucleotides. The binding affinity of the oligonucleotides modified with GuNA[Me]-A, -G, or -mC toward the complementary single-stranded DNAs or RNAs was systematically evaluated. All of the GuNA[Me]-modified oligonucleotides were found to have a strong affinity for RNAs. These data indicate that GuNA[Me] could be a useful modification for therapeutic antisense oligonucleotides.

Graphical Abstract
Introduction
The efficacy and safety of therapeutic oligonucleotides can be controlled by chemical modifications. For applications in antisense technology, chemical modifications aimed at enhancing the duplex-forming ability toward a target RNA (i.e., a complementary single-stranded RNA) and improving the stability against enzymatic degradations are commonly utilized. For instance, antisense oligonucleotides (ASOs) modified with 2',4'-bridged nucleic acid/locked nucleic acid (2',4'-BNA/LNA; Figure 1) are now widely used for gene regulation in vitro and in vivo because 2',4'-BNA/LNA greatly increases the affinity toward the target RNAs, thus enhancing the efficacy of the modified ASOs [1-6]. Notably, the biophysical and pharmacological properties of 2',4'-BNA/LNA-modified ASOs can be further altered with subtle structural changes. Seth and co-workers developed the S-2',4'-constrained-2'-O-ethyl (S-cEt; Figure 1) derivative, which has an exocyclic methyl group in its bridged structure [7,8]. The S-cEt-modified ASOs displayed a higher nuclease resistance and lower hepatotoxicity in in vivo experiments than the corresponding 2',4'-BNA/LNA-modified ASOs [9]; the reduction in hepatotoxicity might be a sequence-dependent phenomenon. Currently, a number of S-cEt-modified ASOs with low hepatotoxicity have been confirmed to be effective for gene regulations in vivo [10,11]. We previously developed amido-bridged nucleic acids (AmNA[R]s) (Figure 1), in which the N-alkyl substituent groups were found to modulate nuclease resistance and hepatic distributions [12]. Wengel’s group reported the synthesis of 2'-amino-LNA (Figure 1) functionalized with a peptide or sugar at the N2'-position, with the aim of modulating the physicochemical properties and specific organ distributions of the therapeutic oligonucleotides [13,14]. A more favorable example is the covalent attachment of a guanidine moiety, which is a common approach to partially neutralize the polyanionic property of oligonucleotides [15-18]. In our previous study, a guanidine-bridged nucleic acid (GuNA[H]; Figure 1) bearing a thymine (T) nucleobase was synthesized as a novel artificial nucleic acid for antisense applications [19]. The modification of oligonucleotides with GuNA[H]-T improved the nuclease resistance, cell membrane permeability, and binding affinity toward complementary single-stranded DNAs (ssDNAs) and RNAs (ssRNAs). We also synthesized and evaluated a GuNA[H]-T analog bearing a methyl group in the guanidine moiety (GuNA[Me]-T; Figure 1) [20]. The GuNA[Me]-T exhibited a similar duplex-forming ability and nuclease resistance as GuNA[H]-T. Since a subtle change in the structure of the 2',4'-BNA/LNA modulated its biophysical and pharmacological properties, in vivo experiments with GuNA[H] and GuNA[Me] are expected to provide further mechanistic insights into how small substituents affect the efficacy and safety of therapeutic oligonucleotides. Thus, the synthesis of GuNA[Me] phosphoramidites bearing other typical nucleobases, i.e., adenine (A), guanine (G), or 5-methylcytosine (mC), instead of the immunologically unfavorable cytosine (C), is needed.
![[1860-5397-17-54-1]](/bjoc/content/figures/1860-5397-17-54-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of 2',4'-BNA/LNA analogs.
Figure 1: Structures of 2',4'-BNA/LNA analogs.
The preparation of all four phosphoramidites (A, G, mC, and T) is generally not easy because each nucleobase differs in the sensitivity to reactions, and appropriate protecting groups need to be selected [8,21-23]. We recently achieved the synthesis of all four GuNA[H] phosphoramidites, where transglycosylations of the 2'-amino-LNA analog with the corresponding nucleobases were performed as the key reactions [24,25]. The transglycosylation is a powerful strategy that simplifies the preparation of phosphoramidites at the late stages of the syntheses [26,27]. Here, we describe the synthesis of GuNA[Me]-A, -G, and -mC phosphoramidites and their incorporations into oligonucleotides. The duplex-forming abilities of all the GuNA[Me]-modified oligonucleotides toward their ssDNA and ssRNA complements were systematically evaluated.
Results and Discussion
Synthesis of the GuNA[Me] phosphoramidites bearing either an A, G, or mC nucleobase
The preparation of the GuNA[Me]-A, -G, and -mC phosphoramidites 3a–c needed for the synthesis of the GuNA[Me]-modified oligonucleotides is detailed in Scheme 1. The acetyl group was selected as a protecting group for the guanidine moiety because it can be easily removed under the basic conditions (ammonia/methylamine solution) used for the DNA synthesis [20]. The phosphoramidite synthesis was started from 2'-amino-LNAs 1a–c, which were rapidly prepared via the transglycosylations of 2'-amino-LNA-T [25]. First, the 2'-amino groups of 1a–c were converted into guanidine moieties with a methyl group using N-acetyl-S,N'-dimethylisothiourea [28], which yielded 65–83% of the products 2a–c. Subsequently, the designed GuNA[Me] phosphoramidites 3a–c were successfully obtained following the phosphitylation of the 3'-hydroxy groups of 2a–c. Notably, since the nucleobases were introduced at the late stage of the synthesis, we had no difficulty preparing these phosphoramidites.
![[1860-5397-17-54-i1]](/bjoc/content/inline/1860-5397-17-54-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The preparation of the GuNA[Me]-A, -G, and -mC phosphoramidites 3a–c. Reagents and conditions: (i) N-acetyl-S,N'-dimethylisothiourea, AgOTf, DIPEA, THF, rt, 72% (2a), 65% (2b), 83% (2c); (ii) (iPr)2NP(Cl)O(CH2)2CN, DIPEA, CH2Cl2, rt, 87% (3a); 65% (3b); 72% (3c).
Scheme 1: The preparation of the GuNA[Me]-A, -G, and -mC phosphoramidites 3a–c. Reagents and conditions: (i) N...
Synthesis of oligonucleotides modified with GuNA[Me]-A, -G, or -mC
The prepared GuNA[Me]-A, -G, and -mC phosphoramidites were incorporated into the middle position of 12-mer oligonucleotides (Table 1). The oligonucleotide synthesis was performed using an automated DNA synthesizer following the established synthetic method for GuNA[Me]-T-modified oligonucleotides [20]. 5-(Ethylthio)-1H-tetrazole (ETT) was used as an activator for the coupling, and the coupling time was extended from 40 s to 20 min for the GuNA[Me] phosphoramidites. Other conditions were the same as those used for general DNA synthesis. After the elongation, the oligonucleotides were treated with ammonia/methylamine solution (7 M NH3 in methanol/40% aqueous methylamine 1:1) at 60 °C for 5 h. Under these conditions, we obtained the GuNA[Me]-mC-modified oligonucleotide ON3 with high purity. In the case of the GuNA[Me] having a purine nucleobase (ON1 and ON2), the acetyl group in the guanidine moiety remained in a considerable amount. This means that we should give attention to the reactivity of each nucleobase. Finally, the acetyl group was successfully removed by extending the deprotection time to 10 h. The yield range of the designed oligonucleotides ON1–ON3 was 12–25%, as shown in Table 1.
Table 1: Synthetic yields and mass spectral data of the GuNA[Me]-modified oligonucleotides ON1–ON3.
| oligonucleotidesa (5'–3') | yield [%] | MALDI–TOF mass | ||
|
found
[M − H]− |
calcd.
[M − H]− |
|||
| d(GCG TTA TTT GCT) | (ON1) | 12 | 3723.9 | 3724.5 |
| d(GCG TTG TTT GCT) | (ON2) | 14 | 3738.9 | 3740.5 |
| d(GCG TTmC TTT GCT) | (ON3) | 25 | 3714.4 | 3714.5 |
aA, G, and mC indicate GuNA[Me] modifications.
Duplex-forming ability of oligonucleotides modified with GuNA[Me]-A, -G, or -mC
The binding affinity of the GuNA[Me]-modified oligonucleotides ON1–ON3 toward ssDNAs or ssRNAs was evaluated by measuring UV melting temperatures (Tm values), and the obtained values were compared with those of the corresponding unmodified oligonucleotides (ON6–ON8). The results are shown in Table 2. As expected, all of the GuNA[Me]-modified oligonucleotides ON1–ON3 exhibited markedly higher Tm values toward ssRNAs than their unmodified counterparts ON6–ON8 (ΔTm = 5–6 °C). These results are similar to those obtained for the GuNA[Me]-T-modified oligonucleotide ON4 (ΔTm = 5 °C). Additionally, the modified ON1–ON3 showed an enhanced duplex-forming ability toward the complementary ssDNAs (ΔTm = 3–6 °C). Among them, GuNA[Me]-A-modified ON1 exhibited a slightly lower ΔTm value than others. This type of nucleobase-dependent difference in ΔTm values is also seen in other GuNA[H]-modified oligonucleotides [25]. Since oligonucleotides modified with 2',4'-BNA/LNA or its analog scpBNA show different nucleobase dependency [1,23], these results could be considered characteristic of the GuNA-modified oligonucleotides. Interactions between the guanidine moiety and nearby base pairing(s) might have affected the ΔTm values, though further investigations are needed for the details.
Table 2: Tm values of duplexes formed between GuNA[Me]-modified oligonucleotides and complementary ssRNAs or ssDNAs.a
| oligonucleotidesa (5'–3') | Tm (ΔTm) [°C] | ||||
| vs ssRNA | vs ssDNA | ||||
| d(GCG TTT TTT GCT)b | (ON5) | 47 | 51 | ||
| d(GCG TTT TTT GCT)b | (ON4) | 52 | (+5) | 56 | (+5) |
| d(GCG TTA TTT GCT) | (ON6) | 45 | 49 | ||
| d(GCG TTA TTT GCT) | (ON1) | 50 | (+5) | 52 | (+3) |
| d(GCG TTG TTT GCT) | (ON7) | 51 | 54 | ||
| d(GCG TTG TTT GCT) | (ON2) | 57 | (+6) | 59 | (+5) |
| d(GCG TTC TTT GCT) | (ON8) | 52 | 53 | ||
| d(GCG TTmC TTT GCT) | (ON3) | 58 | (+6) | 59 | (+6) |
aConditions: 10 mM sodium phosphate buffer (pH 7.2), 100 mM NaCl, 4 µM each oligonucleotide, 0.5 °C/min at 260 nm. Sequences of the complementary ssRNA and ssDNA are 5'-r(AGC AAA NAA CGC)-3' and 5'-d(AGC AAA NAA CGC)-3', respectively. T, A, G, and mC indicate GuNA[Me] modifications. bSee reference [20].
CD spectral analyses of duplexes modified with GuNA[Me]-G
To analyze the structures of the duplexes containing GuNA[Me], circular dichroism (CD) spectra were measured for ON2/ssRNA and ON2/ssDNA duplexes (Figure 2). The CD spectra of ON2/ssRNA and ON2/ssDNA were found to be similar to those of the ON7/ssRNA and ON7/ssDNA duplexes, demonstrating that one modification with GuNA[Me] does not affect the whole duplex structures. Similar results were observed for ON4/ssRNA and ON4/ssDNA (Figure S15 in Supporting Information File 1). In our previous studies, DNA/RNA (A-form) duplexes containing a multiple GuNA[H] modification displayed similar spectral patterns to the natural and the 2',4'-BNA/LNA-modified counterparts [19]. Since GuNA[Me] showed similar results to GuNA[H] in terms of the duplex-forming ability [25], a multiple GuNA[Me] modification to A-form duplexes is also believed not to affect the structures.
![[1860-5397-17-54-2]](/bjoc/content/figures/1860-5397-17-54-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The CD spectra of the ON7/ssRNA, ON2/ssRNA, ON7/ssDNA, and ON2/ssDNA duplexes. Conditions: 10 mM sodium phosphate buffer (pH 7.2), 100 mM NaCl, 4 µM each oligonucleotide. Sequences of the complementary ssRNA and ssDNA are 5'-r(AGC AAA CAA CGC)-3' and 5'-d(AGC AAA CAA CGC)-3', respectively.
Figure 2: The CD spectra of the ON7/ssRNA, ON2/ssRNA, ON7/ssDNA, and ON2/ssDNA duplexes. Conditions: 10 mM so...
Conclusion
We successfully synthesized GuNA[Me] phosphoramidites bearing either an A, G, or mC nucleobase. Each monomer was derived from the corresponding 2'-amino-LNA in two steps and introduced into oligonucleotides. By protecting the guanidine moieties with an acetyl group, we could obtain the oligonucleotides within a 12–25% yield range under the basic conditions (ammonia/methylamine solution) commonly used in oligonucleotide synthesis. The synthesized GuNA[Me]-modified oligonucleotides showed a high binding affinity toward the complementary ssRNAs and ssDNAs. Considering the facile synthesis of the GuNA[Me] monomers and the ability of the GuNA[Me]-modified oligonucleotides to form stable duplexes with ssRNAs, we expect that a modification using GuNA[Me] could be useful for antisense applications. In our ongoing studies, we are evaluating the efficacy of ASOs modified with GuNAs in vitro and in vivo, and the results will be reported in due course.
Experimental
Chemicals and instrumentation
All moisture-sensitive reactions were carried out in well-dried glassware under N2 atmosphere. Dehydrated acetonitrile, dichloromethane, and tetrahydrofuran (THF) were used as purchased. 1H, 13C, and 31P NMR spectra were recorded using a JEOL JNM-ECS300 spectrometer. The chemical shift values are expressed in δ values (ppm) relative to tetramethylsilane as an internal standard, CHCl3 (δ = 7.26 ppm) for 1H NMR, CDCl3 (δ = 77.0 ppm) for 13C NMR, and 5% H3PO4 (δ = 0 ppm) for 31P NMR. Infrared (IR) spectra were recorded using a JASCO FT/IR-4200 spectrometer. The optical rotation was recorded using a JASCO P-2200 instrument. A MALDI–TOF mass spectrometer (SpiralTOF JMS-S3000) was used to measure the mass spectra of all compounds. For column chromatography, silica gel PSQ 60B or 100B was used. The progress of the reactions was monitored by analytical thin-layer chromatography on glass plates (TLC Silica gel 60 F254), and the products were visualized using UV light.
Synthesis of phosphoramidites
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-3-(N6-benzoyladenine-9-yl)-1-(4,4'-dimethoxytrityl)oxymethyl-2-oxa-5-azabicyclo[2.2.1]heptan-7-ol (2a): This compound was synthesized in a similar manner as described in reference [20]. To the mixture of compound 1a (841 mg, 1.23 mmol) and N-acetyl-S,N'-dimethylisothiourea (271 mg, 1.84 mmol), anhydrous THF (12 mL) was added and the mixture placed in an ice bath under stirring. Subsequently, N,N-diisopropylethylamine (0.35 mL, 2.0 mmol) and silver triflate (507 mg, 1.97 mmol) were added, and the mixture was stirred at room temperature overnight. Upon completion of the reaction, the mixture was diluted with ethyl acetate, after which sat. aq. NaCl was added. Following filtration, the product was extracted with ethyl acetate, washed with water and brine, dried (using Na2SO4), and concentrated. The product was then purified using column chromatography to yield 2a (698 mg, 72%) as a yellow solid substance. 2a: ![[Graphic 1]](/bjoc/content/inline/1860-5397-17-54-i2.svg?max-width=637&scale=1.18182) −26.4 (c 1.0, CHCl3); IR (KBr): 2999, 2952, 2837, 1696, 1606, 1509, 1451, 1410, 1297, 1251, 1177, 1155, 1074, 1035 cm−1; 1H NMR (CDCl3) δ 2.00 (s, 3H), 2.80 (s, 3H), 3.48, 3.57 (AB, J = 10.7 Hz, 2H), 3.67 (s, 2H), 3.74 (s, 3H), 3.74 (s, 3H), 4.37 (s, 1H), 5.02 (s, 1H), 6.14 (s1H), 6.79 ( d, J = 8.9 Hz, 4H), 7.15–7.33 (m, 7H), 7.42–7.61 (m, 5H), 7.98 (dd, J = 1.4 Hz, 7.2 Hz, 2H), 8.25 (s, 1H), 8.70 (s, 1H), 9.15 (s, 1H); 13C NMR (CDCl3) δ 26.44, 29.70, 54.23, 55.16, 60.27, 63.88, 71.06, 85.51, 86.46, 88.50, 113.18, 123.47, 126.95, 127.86, 127.90, 128.03, 128.84, 129.94, 130.01, 132.87, 133.31, 135.21, 135.47, 140.48, 144.26, 149.38, 150.80, 152.55, 158.52, 164.76; HRMS–MALDI (m/z): [M + Na]+ calcd for C43H42N8O7Na, 805.3069; found, 805.3063.
−26.4 (c 1.0, CHCl3); IR (KBr): 2999, 2952, 2837, 1696, 1606, 1509, 1451, 1410, 1297, 1251, 1177, 1155, 1074, 1035 cm−1; 1H NMR (CDCl3) δ 2.00 (s, 3H), 2.80 (s, 3H), 3.48, 3.57 (AB, J = 10.7 Hz, 2H), 3.67 (s, 2H), 3.74 (s, 3H), 3.74 (s, 3H), 4.37 (s, 1H), 5.02 (s, 1H), 6.14 (s1H), 6.79 ( d, J = 8.9 Hz, 4H), 7.15–7.33 (m, 7H), 7.42–7.61 (m, 5H), 7.98 (dd, J = 1.4 Hz, 7.2 Hz, 2H), 8.25 (s, 1H), 8.70 (s, 1H), 9.15 (s, 1H); 13C NMR (CDCl3) δ 26.44, 29.70, 54.23, 55.16, 60.27, 63.88, 71.06, 85.51, 86.46, 88.50, 113.18, 123.47, 126.95, 127.86, 127.90, 128.03, 128.84, 129.94, 130.01, 132.87, 133.31, 135.21, 135.47, 140.48, 144.26, 149.38, 150.80, 152.55, 158.52, 164.76; HRMS–MALDI (m/z): [M + Na]+ calcd for C43H42N8O7Na, 805.3069; found, 805.3063.
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-1-(4,4'-dimethoxytrityl)oxymethyl-3-(O6-diphenylcarbamoyl-N2-isobutyrylguanine-9-yl)-2-oxa-5-azabicyclo[2.2.1]heptan-7-ol (2b): This compound was synthesized in a similar manner as described in reference [20]. To the mixture of compound 1b (2.15 g, 2.49 mmol) and N-acetyl-S,N'-dimethylisothiourea (402 mg, 2.75 mmol), anhydrous THF (25 mL) was added and the mixture placed in an ice bath under stirring. Subsequently, N,N-diisopropylethylamine (0.57 mL, 3.3 mmol) and silver triflate (835 mg, 3.25 mmol) were added, and the mixture was stirred at room temperature for 2 h. Upon completion of the reaction, the mixture was diluted with ethyl acetate and washed with sat. aq. NaHCO3, after which sat. aq. NH4Cl was added. Following filtration, the product was extracted with ethyl acetate, washed with water and brine, dried (using Na2SO4), and concentrated. The product was purified using column chromatography to yield 2b (1.56 g, 65%) as a yellow solid substance. 2b: ![[Graphic 2]](/bjoc/content/inline/1860-5397-17-54-i3.svg?max-width=637&scale=1.18182) −15.2 (c 1.0, CHCl3); IR (KBr): 3350, 2971, 2837, 1750, 1712, 1587, 1509, 1444, 1411, 1335, 1284, 1249, 1226, 1176, 1116, 1068, 1035 cm−1; 1H NMR (CDCl3) δ 1.23 (d, J = 6.5 Hz, 3H), 1.25 (d, J = 6.2 Hz, 3H), 2.05 (s, 3H), 2.55–2.66 (m, 1H), 3.05 (d, J = 4.2 Hz, 3H), 3.48, 3.53 (AB, J = 10.8 Hz, 2H), 3.59, 3.74 (AB, J = 10.3 Hz, 2H), 3.78 (s, 3H), 3.78 (s, 3H), 4.29 (s, 1H), 5.07 (s, 1H), 6.00 (s, 1H), 6.83 (d, J = 7.9 Hz, 4H), 7.16–7.45 (m, 18H), 8.13 (s, 1H), 8.16 (s, 1H); 13C NMR (CDCl3) δ 19.19, 19.34, 26.03, 29.96, 36.61, 54.14, 55.14, 60.15, 63.64, 72.12, 85.49, 86.35, 88.17, 113.19, 121.57, 126.91, 127.90, 128.02, 129.18, 129.94, 129.99, 135.31, 135.52, 141.55, 144.34, 150.26, 151.60, 153.12, 156.07, 158.50, 162.61, 174.91; HRMS (MALDI) (m/z): [M + Na]+ calcd. for C53H53N9O9Na, 982.3858; found, 982.3856.
−15.2 (c 1.0, CHCl3); IR (KBr): 3350, 2971, 2837, 1750, 1712, 1587, 1509, 1444, 1411, 1335, 1284, 1249, 1226, 1176, 1116, 1068, 1035 cm−1; 1H NMR (CDCl3) δ 1.23 (d, J = 6.5 Hz, 3H), 1.25 (d, J = 6.2 Hz, 3H), 2.05 (s, 3H), 2.55–2.66 (m, 1H), 3.05 (d, J = 4.2 Hz, 3H), 3.48, 3.53 (AB, J = 10.8 Hz, 2H), 3.59, 3.74 (AB, J = 10.3 Hz, 2H), 3.78 (s, 3H), 3.78 (s, 3H), 4.29 (s, 1H), 5.07 (s, 1H), 6.00 (s, 1H), 6.83 (d, J = 7.9 Hz, 4H), 7.16–7.45 (m, 18H), 8.13 (s, 1H), 8.16 (s, 1H); 13C NMR (CDCl3) δ 19.19, 19.34, 26.03, 29.96, 36.61, 54.14, 55.14, 60.15, 63.64, 72.12, 85.49, 86.35, 88.17, 113.19, 121.57, 126.91, 127.90, 128.02, 129.18, 129.94, 129.99, 135.31, 135.52, 141.55, 144.34, 150.26, 151.60, 153.12, 156.07, 158.50, 162.61, 174.91; HRMS (MALDI) (m/z): [M + Na]+ calcd. for C53H53N9O9Na, 982.3858; found, 982.3856.
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-1-(4,4'-dimethoxytrityl)oxymethyl-3-(O6-diphenylcarbamoyl-N2-isobutyrylguanine-9-yl)-2-oxa-5-azabicyclo[2.2.1]heptan-7-ol (2c): This compound was synthesized in a similar manner as described in reference [20]. To the mixture of compound 1c (679 mg, 1.01 mmol) and N-acetyl-S,N'-dimethylisothiourea (194 mg, 1.33 mmol), anhydrous THF (10 mL) was added and the mixture placed in an ice bath under stirring. Subsequently, N,N-diisopropylethylamine (0.28 mL, 1.6 mmol) and silver triflate (411 mg, 1.60 mmol) were added, and the mixture was stirred at room temperature for 1 h. Upon completion of the reaction, the mixture was diluted with ethyl acetate, after which sat. aq. NH4Cl was added. Following filtration, the product was extracted with ethyl acetate, washed with water and brine, dried (using Na2SO4), and concentrated. The product was purified using column chromatography to yield 2c (641 mg, 83%) as a white solid substance. 2c: 1H NMR (CDCl3) δ 1.82 (s, 3H), 2.03 (d, J = 4.2 Hz, 3H), 2.81 (s, 3H), 3.31, 3.51 (AB, J = 9.7 Hz, 2H), 3.48, 3.57 (AB, J = 10.9 Hz, 2H), 3.76 (s, 3H), 3.77 (s, 3H), 4.31 (s, 1H), 4.60 (s, 1H), 5.56 (s, 1H), 6.83 (dd, J = 8.6 Hz, 1.7 Hz, 4H), 7.21–7.55 (m, 12H), 7.72 (s, 1H), 8.30 (d, J = 7.2 Hz, 2H); 13C NMR (CDCl3) δ 13.62, 26.30, 29.47, 53.81, 54.04, 55.17, 58.97, 63.18, 69.93, 86.08, 86.66, 88.70, 111.89, 113.24, 127.03, 128.00, 128.07, 129.86, 129.94, 130.11, 132.52, 135.14, 135.57, 136.87, 144.23, 147.92, 158.59, 159.66, 161.16, 179.48; HRMS–MALDI (m/z): [M + Na]+ calcd for C43H44N6O8Na, 795.3113; found, 795.3106.
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-3-(N6-benzoyladenine-9-yl)-7-[2-cyanoethoxy(diisopropylamino)phosphanyl]oxyl-1-(4,4'-dimethoxytrityl)oxymethyl-2-oxa-5-azabicyclo[2.2.1]heptane (3a): This compound was synthesized in a similar manner as described in reference [20]. To a solution of 2a (1.47 g, 1.9 mmol) in dichloromethane (19 mL), N,N-diisopropylethylamine (0.7 mL, 4.1 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.8 mL, 3.8 mmol) were added, and the mixture was stirred at room temperature for 6 h. Upon completion of the reaction, sat. aq. NaHCO3 was added, and the product was extracted with dichloromethane. The organic phase was washed with water and brine, dried (using Na2SO4), and concentrated. The product was purified using column chromatography to yield 3a (1.62 g, 87%) as a yellow solid substance. 3a: 31P NMR (CDCl3) δ 149.15, 149.31; HRMS–MALDI (m/z): [M + Na]+ calcd for C52H59N10O8NaP, 1005.4147; found, 1005.4143.
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-7-[2-cyanoethoxy(diisopropylamino)phosphanyl]oxyl-1-(4,4'-dimethoxytrityl)oxymethyl-3-(O6-diphenylcarbamoyl-N2-isobutyrylguanine-9-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (3b): This compound was synthesized in a similar manner as described in reference [20]. To a solution of 2b (149 mg, 0.155 mmol) in dichloromethane (1.5 mL), N,N-diisopropylethylamine (56 µL, 0.32 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (69 µL, 0.31 mmol) were added, and the mixture was stirred at room temperature for 6 h. Upon completion of the reaction, sat. aq. NaHCO3 was added, and the product was extracted with dichloromethane. The organic phase was washed with water and brine, dried (using Na2SO4), and concentrated. The product was purified using column chromatography to yield 3b (117 mg, 65%) as a yellow solid substance. 3b: 31P NMR (CDCl3) δ 148.80, 149.55; HRMS–MALDI (m/z): [M + Na]+ calcd for C62H70N11O10NaP, 1182.4937; found, 1182.4955.
(1R,3R,4R,7S)-5-(N'-Acetyl-N-methylcarbamimidoyl)-7-[2-cyanoethoxy(diisopropylamino)phosphanyl]oxyl-1-(4,4'-dimethoxytrityl)oxymethyl-3-(O6-diphenylcarbamoyl-N2-isobutyrylguanine-9-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (3c): This compound was synthesized in a similar manner as described in reference [20]. To a solution of 2c (1.08 g, 1.40 mmol) in dichloromethane (14 mL), N,N-diisopropylethylamine (0.8 mL, 4.3 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.6 mL, 2.8 mmol) were added, and the mixture was stirred at room temperature for 6 h. Upon completion of the reaction, sat. aq. NaHCO3 was added, and the product was extracted with dichloromethane. The organic phase was washed with water and brine, dried (using Na2SO4), and concentrated. The product was purified using column chromatography to yield 3c (0.98 g, 72%) as a yellow solid substance. 3c: 31P NMR (CDCl3) δ 148.61, 148.85; HRMS–MALDI (m/z): [M + Na]+ calcd for C52H61N8O9NaP, 995.4191; found, 995.4181.
Oligonucleotide synthesis and purification
The synthesis of the oligonucleotides modified with GuNA[Me]-A, -G, or -mC (0.2 µmol scale) was performed using the nS-8 oligonucleotide synthesizer (GeneDesign, Inc.) according to the standard phosphoramidite protocol with 0.5 M 5-ethylthiotetrazole as an activator. The protocol is similar to that described in reference [20]. A Custom Primer Support™ T 40s (GE Healthcare) was used as a solid support. The amidite solution was dehydrated. The standard synthesis cycle was used for the assembly of the reagents except that the coupling time was extended to 16 min. The synthesis was carried out in the trityl-on mode. The oligonucleotides were treated with a 1:1 mixture of 7 N ammonia solution in methanol and 40% aq. methylamine at room temperature for 10 h to remove the solid support, and then the mixture was heated at 60 °C for 10 h (mC) or 15 h (A and G). After deprotection, the oligonucleotides were rapidly purified using a Sep-Pac® Plus C18 Cartridge. Subsequently, the desired oligonucleotides were further purified using reversed-phase HPLC with Waters XBridge™ C18 (4.6 × 50 mm analytical and 10 mm × 50 mm preparative) columns, with a linear gradient of MeCN (2.5–5% over 5 min, then 5–7.5% over 20 min) in 0.1 M triethylammonium acetate buffer (pH 7.0). The purity and structure of the oligonucleotides were confirmed by HPLC and MALDI–TOF mass spectrometry, respectively.
UV melting experiments and melting profiles
The UV melting experiments were carried out using SHIMADZU UV-1650PC and SHIMADZU UV-1800 spectrometers equipped with a Tm analysis accessory. Equimolecular amounts of the target ssRNAs or ssDNAs and the oligonucleotides were dissolved in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl to achieve a final strand concentration of 4 µM. The samples were annealed by heating at 95 °C followed by slow cooling to room temperature. The melting profile was recorded at 260 nm from 0 to 90 °C at a scan rate of 0.5 °C/min. The Tm values were taken as the temperatures at which the formed duplexes were half dissociated, determined by the midline of the melting curves.
CD spectrum measurement
The CD spectra were recorded at 10 °C in a quartz cuvette of 1 cm optical path length. The samples were prepared in the same manner as described in the UV melting experiments. The molar ellipticity was calculated from the equation [θ] = θ/cl, where θ, c, and l indicate the relative intensity, sample concentration, and path length in centimeters, respectively.
Supporting Information
| Supporting Information File 1: 1H, 13C, and 32P NMR spectra for all new compounds, HPLC charts and MALDI–TOF mass data for all new oligonucleotides, UV melting curves of the duplexes formed between GuNA[Me]-modified oligonucleotides and ssDNAs (or ssRNAs), and CD spectra of ON4/ssRNA and ON4/ssDNA. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Kaur, H.; Babu, B. R.; Maiti, S. Chem. Rev. 2007, 107, 4672–4697. doi:10.1021/cr050266u
Return to citation in text: [1] [2] -
Amodio, N.; Stamato, M. A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M. E. G.; Romeo, E.; Raimondi, L.; Caracciolo, D.; Zuccalà, V.; Rossi, M.; Neri, A.; Munshi, N. C.; Tagliaferri, P.; Tassone, P. Leukemia 2018, 32, 1948–1957. doi:10.1038/s41375-018-0067-3
Return to citation in text: [1] -
Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Clin. Cancer Res. 2019, 25, 7189–7201. doi:10.1158/1078-0432.ccr-17-3024
Return to citation in text: [1] -
Chery, J.; Petri, A.; Wagschal, A.; Lim, S.-Y.; Cunningham, J.; Vasudevan, S.; Kauppinen, S.; Näär, A. M. Nucleic Acid Ther. 2018, 28, 273–284. doi:10.1089/nat.2018.0722
Return to citation in text: [1] -
Javanbakht, H.; Mueller, H.; Walther, J.; Zhou, X.; Lopez, A.; Pattupara, T.; Blaising, J.; Pedersen, L.; Albæk, N.; Jackerott, M.; Shi, T.; Ploix, C.; Driessen, W.; Persson, R.; Ravn, J.; Young, J. A. T.; Ottosen, S. Mol. Ther.–Nucleic Acids 2018, 11, 441–454. doi:10.1016/j.omtn.2018.02.005
Return to citation in text: [1] -
Shimo, T.; Tachibana, K.; Saito, K.; Yoshida, T.; Tomita, E.; Waki, R.; Yamamoto, T.; Doi, T.; Inoue, T.; Kawakami, J.; Obika, S. Nucleic Acids Res. 2014, 42, 8174–8187. doi:10.1093/nar/gku512
Return to citation in text: [1] -
Seth, P. P.; Siwkowski, A.; Allerson, C. R.; Vasquez, G.; Lee, S.; Prakash, T. P.; Kinberger, G.; Migawa, M. T.; Gaus, H.; Bhat, B.; Swayze, E. E. Nucleic Acids Symp. Ser. 2008, 52, 553–554. doi:10.1093/nass/nrn280
Return to citation in text: [1] -
Seth, P. P.; Vasquez, G.; Allerson, C. A.; Berdeja, A.; Gaus, H.; Kinberger, G. A.; Prakash, T. P.; Migawa, M. T.; Bhat, B.; Swayze, E. E. J. Org. Chem. 2010, 75, 1569–1581. doi:10.1021/jo902560f
Return to citation in text: [1] [2] -
Seth, P. P.; Siwkowski, A.; Allerson, C. R.; Vasquez, G.; Lee, S.; Prakash, T. P.; Wancewicz, E. V.; Witchell, D.; Swayze, E. E. J. Med. Chem. 2009, 52, 10–13. doi:10.1021/jm801294h
Return to citation in text: [1] -
Carroll, J. B.; Warby, S. C.; Southwell, A. L.; Doty, C. N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C. F.; Freier, S. M.; Hayden, M. R. Mol. Ther. 2011, 19, 2178–2185. doi:10.1038/mt.2011.201
Return to citation in text: [1] -
Pandey, S. K.; Wheeler, T. M.; Justice, S. L.; Kim, A.; Younis, H. S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E. E.; Freier, S. M.; Bennett, C. F.; Thornton, C. A.; MacLeod, A. R. J. Pharmacol. Exp. Ther. 2015, 355, 329–340. doi:10.1124/jpet.115.226969
Return to citation in text: [1] -
Yamamoto, T.; Yahara, A.; Waki, R.; Yasuhara, H.; Wada, F.; Harada-Shiba, M.; Obika, S. Org. Biomol. Chem. 2015, 13, 3757–3765. doi:10.1039/c5ob00242g
Return to citation in text: [1] -
Johannsen, M. W.; Crispino, L.; Wamberg, M. C.; Kalra, N.; Wengel, J. Org. Biomol. Chem. 2011, 9, 243–252. doi:10.1039/c0ob00532k
Return to citation in text: [1] -
Kumar, R.; Ries, A.; Wengel, J. Molecules 2017, 22, 852. doi:10.3390/molecules22050852
Return to citation in text: [1] -
Prakash, T. P.; Püschl, A.; Lesnik, E.; Mohan, V.; Tereshko, V.; Egli, M.; Manoharan, M. Org. Lett. 2004, 6, 1971–1974. doi:10.1021/ol049470e
Return to citation in text: [1] -
Brzezinska, J.; D’Onofrio, J.; Buff, M. C. R.; Hean, J.; Ely, A.; Marimani, M.; Arbuthnot, P.; Engels, J. W. Bioorg. Med. Chem. 2012, 20, 1594–1606. doi:10.1016/j.bmc.2011.12.024
Return to citation in text: [1] -
Deglane, G.; Abes, S.; Michel, T.; Prévot, P.; Vives, E.; Debart, F.; Barvik, I.; Lebleu, B.; Vasseur, J.-J. ChemBioChem 2006, 7, 684–692. doi:10.1002/cbic.200500433
Return to citation in text: [1] -
Barman, J.; Gurav, D.; Oommen, O. P.; Varghese, O. P. RSC Adv. 2015, 5, 12257–12260. doi:10.1039/c4ra14721a
Return to citation in text: [1] -
Shrestha, A. R.; Kotobuki, Y.; Hari, Y.; Obika, S. Chem. Commun. 2014, 50, 575–577. doi:10.1039/c3cc46017g
Return to citation in text: [1] [2] -
Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Koshkin, A. A.; Singh, S. K.; Nielsen, P.; Rajwanshi, V. K.; Kumar, R.; Meldgaard, M.; Olsen, C. E.; Wengel, J. Tetrahedron 1998, 54, 3607–3630. doi:10.1016/s0040-4020(98)00094-5
Return to citation in text: [1] -
Mitsuoka, Y.; Kodama, T.; Ohnishi, R.; Hari, Y.; Imanishi, T.; Obika, S. Nucleic Acids Res. 2009, 37, 1225–1238. doi:10.1093/nar/gkn1062
Return to citation in text: [1] -
Horiba, M.; Yamaguchi, T.; Obika, S. J. Org. Chem. 2016, 81, 11000–11008. doi:10.1021/acs.joc.6b02036
Return to citation in text: [1] [2] -
Sawamoto, H.; Arai, Y.; Yamakoshi, S.; Obika, S.; Kawanishi, E. Org. Lett. 2018, 20, 1928–1931. doi:10.1021/acs.orglett.8b00476
Return to citation in text: [1] -
Kumagai, S.; Sawamoto, H.; Takegawa-Araki, T.; Arai, Y.; Yamakoshi, S.; Yamada, K.; Ohta, T.; Kawanishi, E.; Horie, N.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2020, 18, 9461–9472. doi:10.1039/d0ob01970d
Return to citation in text: [1] [2] [3] [4] -
Umemoto, T.; Masada, S.; Miyata, K.; Ogasawara-Shimizu, M.; Murata, S.; Nishi, K.; Ogi, K.; Hayase, Y.; Cho, N. Tetrahedron 2017, 73, 1211–1218. doi:10.1016/j.tet.2017.01.010
Return to citation in text: [1] -
Fujisaka, A.; Hari, Y.; Takuma, H.; Rahman, S. M. A.; Yoshikawa, H.; Pang, J.; Imanishi, T.; Obika, S. Bioorg. Med. Chem. 2019, 27, 1728–1741. doi:10.1016/j.bmc.2019.02.034
Return to citation in text: [1] -
Michael, J. D.; Ross, B. C.; Rees, P. M. Tetrahedron Lett. 1985, 26, 4149–4152. doi:10.1016/s0040-4039(00)89316-8
Return to citation in text: [1]
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 1. | Kaur, H.; Babu, B. R.; Maiti, S. Chem. Rev. 2007, 107, 4672–4697. doi:10.1021/cr050266u |
| 2. | Amodio, N.; Stamato, M. A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M. E. G.; Romeo, E.; Raimondi, L.; Caracciolo, D.; Zuccalà, V.; Rossi, M.; Neri, A.; Munshi, N. C.; Tagliaferri, P.; Tassone, P. Leukemia 2018, 32, 1948–1957. doi:10.1038/s41375-018-0067-3 |
| 3. | Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Clin. Cancer Res. 2019, 25, 7189–7201. doi:10.1158/1078-0432.ccr-17-3024 |
| 4. | Chery, J.; Petri, A.; Wagschal, A.; Lim, S.-Y.; Cunningham, J.; Vasudevan, S.; Kauppinen, S.; Näär, A. M. Nucleic Acid Ther. 2018, 28, 273–284. doi:10.1089/nat.2018.0722 |
| 5. | Javanbakht, H.; Mueller, H.; Walther, J.; Zhou, X.; Lopez, A.; Pattupara, T.; Blaising, J.; Pedersen, L.; Albæk, N.; Jackerott, M.; Shi, T.; Ploix, C.; Driessen, W.; Persson, R.; Ravn, J.; Young, J. A. T.; Ottosen, S. Mol. Ther.–Nucleic Acids 2018, 11, 441–454. doi:10.1016/j.omtn.2018.02.005 |
| 6. | Shimo, T.; Tachibana, K.; Saito, K.; Yoshida, T.; Tomita, E.; Waki, R.; Yamamoto, T.; Doi, T.; Inoue, T.; Kawakami, J.; Obika, S. Nucleic Acids Res. 2014, 42, 8174–8187. doi:10.1093/nar/gku512 |
| 12. | Yamamoto, T.; Yahara, A.; Waki, R.; Yasuhara, H.; Wada, F.; Harada-Shiba, M.; Obika, S. Org. Biomol. Chem. 2015, 13, 3757–3765. doi:10.1039/c5ob00242g |
| 28. | Michael, J. D.; Ross, B. C.; Rees, P. M. Tetrahedron Lett. 1985, 26, 4149–4152. doi:10.1016/s0040-4039(00)89316-8 |
| 10. | Carroll, J. B.; Warby, S. C.; Southwell, A. L.; Doty, C. N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C. F.; Freier, S. M.; Hayden, M. R. Mol. Ther. 2011, 19, 2178–2185. doi:10.1038/mt.2011.201 |
| 11. | Pandey, S. K.; Wheeler, T. M.; Justice, S. L.; Kim, A.; Younis, H. S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E. E.; Freier, S. M.; Bennett, C. F.; Thornton, C. A.; MacLeod, A. R. J. Pharmacol. Exp. Ther. 2015, 355, 329–340. doi:10.1124/jpet.115.226969 |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 9. | Seth, P. P.; Siwkowski, A.; Allerson, C. R.; Vasquez, G.; Lee, S.; Prakash, T. P.; Wancewicz, E. V.; Witchell, D.; Swayze, E. E. J. Med. Chem. 2009, 52, 10–13. doi:10.1021/jm801294h |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 7. | Seth, P. P.; Siwkowski, A.; Allerson, C. R.; Vasquez, G.; Lee, S.; Prakash, T. P.; Kinberger, G.; Migawa, M. T.; Gaus, H.; Bhat, B.; Swayze, E. E. Nucleic Acids Symp. Ser. 2008, 52, 553–554. doi:10.1093/nass/nrn280 |
| 8. | Seth, P. P.; Vasquez, G.; Allerson, C. A.; Berdeja, A.; Gaus, H.; Kinberger, G. A.; Prakash, T. P.; Migawa, M. T.; Bhat, B.; Swayze, E. E. J. Org. Chem. 2010, 75, 1569–1581. doi:10.1021/jo902560f |
| 25. | Kumagai, S.; Sawamoto, H.; Takegawa-Araki, T.; Arai, Y.; Yamakoshi, S.; Yamada, K.; Ohta, T.; Kawanishi, E.; Horie, N.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2020, 18, 9461–9472. doi:10.1039/d0ob01970d |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 24. | Sawamoto, H.; Arai, Y.; Yamakoshi, S.; Obika, S.; Kawanishi, E. Org. Lett. 2018, 20, 1928–1931. doi:10.1021/acs.orglett.8b00476 |
| 25. | Kumagai, S.; Sawamoto, H.; Takegawa-Araki, T.; Arai, Y.; Yamakoshi, S.; Yamada, K.; Ohta, T.; Kawanishi, E.; Horie, N.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2020, 18, 9461–9472. doi:10.1039/d0ob01970d |
| 19. | Shrestha, A. R.; Kotobuki, Y.; Hari, Y.; Obika, S. Chem. Commun. 2014, 50, 575–577. doi:10.1039/c3cc46017g |
| 26. | Umemoto, T.; Masada, S.; Miyata, K.; Ogasawara-Shimizu, M.; Murata, S.; Nishi, K.; Ogi, K.; Hayase, Y.; Cho, N. Tetrahedron 2017, 73, 1211–1218. doi:10.1016/j.tet.2017.01.010 |
| 27. | Fujisaka, A.; Hari, Y.; Takuma, H.; Rahman, S. M. A.; Yoshikawa, H.; Pang, J.; Imanishi, T.; Obika, S. Bioorg. Med. Chem. 2019, 27, 1728–1741. doi:10.1016/j.bmc.2019.02.034 |
| 15. | Prakash, T. P.; Püschl, A.; Lesnik, E.; Mohan, V.; Tereshko, V.; Egli, M.; Manoharan, M. Org. Lett. 2004, 6, 1971–1974. doi:10.1021/ol049470e |
| 16. | Brzezinska, J.; D’Onofrio, J.; Buff, M. C. R.; Hean, J.; Ely, A.; Marimani, M.; Arbuthnot, P.; Engels, J. W. Bioorg. Med. Chem. 2012, 20, 1594–1606. doi:10.1016/j.bmc.2011.12.024 |
| 17. | Deglane, G.; Abes, S.; Michel, T.; Prévot, P.; Vives, E.; Debart, F.; Barvik, I.; Lebleu, B.; Vasseur, J.-J. ChemBioChem 2006, 7, 684–692. doi:10.1002/cbic.200500433 |
| 18. | Barman, J.; Gurav, D.; Oommen, O. P.; Varghese, O. P. RSC Adv. 2015, 5, 12257–12260. doi:10.1039/c4ra14721a |
| 13. | Johannsen, M. W.; Crispino, L.; Wamberg, M. C.; Kalra, N.; Wengel, J. Org. Biomol. Chem. 2011, 9, 243–252. doi:10.1039/c0ob00532k |
| 14. | Kumar, R.; Ries, A.; Wengel, J. Molecules 2017, 22, 852. doi:10.3390/molecules22050852 |
| 8. | Seth, P. P.; Vasquez, G.; Allerson, C. A.; Berdeja, A.; Gaus, H.; Kinberger, G. A.; Prakash, T. P.; Migawa, M. T.; Bhat, B.; Swayze, E. E. J. Org. Chem. 2010, 75, 1569–1581. doi:10.1021/jo902560f |
| 21. | Koshkin, A. A.; Singh, S. K.; Nielsen, P.; Rajwanshi, V. K.; Kumar, R.; Meldgaard, M.; Olsen, C. E.; Wengel, J. Tetrahedron 1998, 54, 3607–3630. doi:10.1016/s0040-4020(98)00094-5 |
| 22. | Mitsuoka, Y.; Kodama, T.; Ohnishi, R.; Hari, Y.; Imanishi, T.; Obika, S. Nucleic Acids Res. 2009, 37, 1225–1238. doi:10.1093/nar/gkn1062 |
| 23. | Horiba, M.; Yamaguchi, T.; Obika, S. J. Org. Chem. 2016, 81, 11000–11008. doi:10.1021/acs.joc.6b02036 |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 25. | Kumagai, S.; Sawamoto, H.; Takegawa-Araki, T.; Arai, Y.; Yamakoshi, S.; Yamada, K.; Ohta, T.; Kawanishi, E.; Horie, N.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2020, 18, 9461–9472. doi:10.1039/d0ob01970d |
| 1. | Kaur, H.; Babu, B. R.; Maiti, S. Chem. Rev. 2007, 107, 4672–4697. doi:10.1021/cr050266u |
| 23. | Horiba, M.; Yamaguchi, T.; Obika, S. J. Org. Chem. 2016, 81, 11000–11008. doi:10.1021/acs.joc.6b02036 |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 20. | Horie, N.; Kumagai, S.; Kotobuki, Y.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2018, 16, 6531–6536. doi:10.1039/c8ob01307a |
| 19. | Shrestha, A. R.; Kotobuki, Y.; Hari, Y.; Obika, S. Chem. Commun. 2014, 50, 575–577. doi:10.1039/c3cc46017g |
| 25. | Kumagai, S.; Sawamoto, H.; Takegawa-Araki, T.; Arai, Y.; Yamakoshi, S.; Yamada, K.; Ohta, T.; Kawanishi, E.; Horie, N.; Yamaguchi, T.; Obika, S. Org. Biomol. Chem. 2020, 18, 9461–9472. doi:10.1039/d0ob01970d |
© 2021 Horie et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)