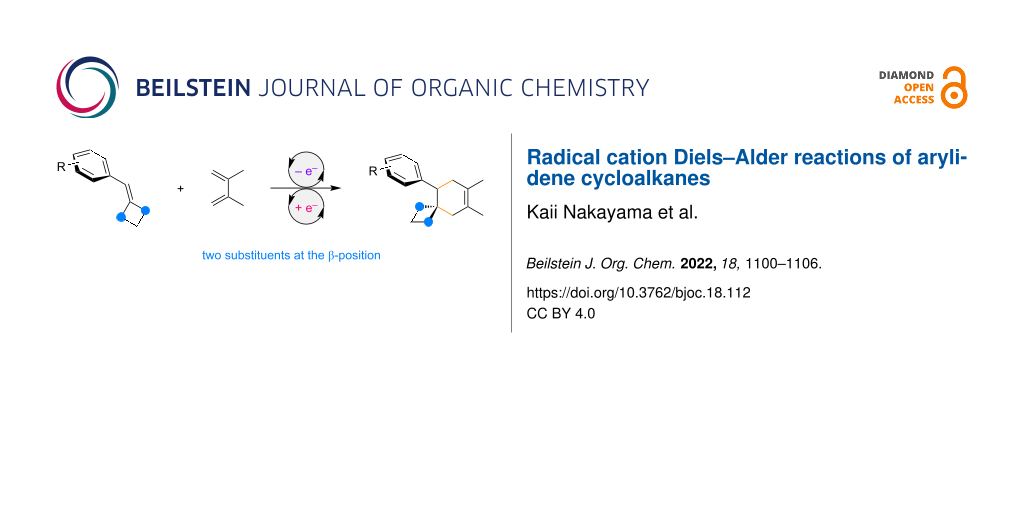
Abstract
TiO2 photoelectrochemical and electrochemical radical cation Diels–Alder reactions of arylidene cycloalkanes are described, leading to the construction of spiro ring systems. Although the mechanism remains an open question, arylidene cyclobutanes are found to be much more effective in the reaction than other cycloalkanes. Since the reaction is completed with a substoichiometric amount of electricity, a radical cation chain pathway is likely to be involved.

Graphical Abstract
Introduction
Single-electron transfer is one of the simplest modes for small molecule activation, employing a polarity inversion to generate radical ions which have proven to be unique reactive intermediates in the field of synthetic organic chemistry. A radical cation Diels–Alder reaction is a typical example of this activation mode since both the original diene and dienophile are electron-rich and thus not an effective combination of reactants [1-11]. Single-electron transfer makes the construction of six-membered ring systems possible. In general, single-electron oxidation of an electron-rich dienophile generates its radical cation which is then trapped by the diene (Figure 1). Since the forming cyclohexene remains in the radical cation state as well, one electron reduction is required to complete the net redox neutral transformation. Therefore, a chain pathway can be involved, where an electron acts as a catalyst rather than a reagent [12-18]. In this reaction format, trans-anethole is an electron-rich dienophile and has widely been studied as a benchmark for single-electron transfer using photochemical and electrochemical methods [19-32]. A one electron oxidant can also be an initiator for this transformation [33-35]. Overall, the scope of the reaction has been expanding. Starting from trans-anethole, several functionalities at the β-position are found to be compatible with the reaction, while those at the aryl ring are somewhat limited (Figure 2). It should be noted that a second substituent at the β-position of trans-anethole has a significant impact on the reaction and surprisingly, even an additional methyl group is not acceptable.
![[1860-5397-18-112-1]](/bjoc/content/figures/1860-5397-18-112-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Plausible mechanism of the radical cation Diels–Alder reaction (EDG: electron-donating group).
Figure 1: Plausible mechanism of the radical cation Diels–Alder reaction (EDG: electron-donating group).
![[1860-5397-18-112-2]](/bjoc/content/figures/1860-5397-18-112-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Landscape of the radical cation Diels–Alder reaction.
Figure 2: Landscape of the radical cation Diels–Alder reaction.
We have developed radical cation cycloadditions using (photo)electrochemical single-electron transfer in lithium perchlorate (LiClO4)/nitromethane (CH3NO2) solution [36-44]. During the course of our studies, we found that the TiO2 photoelectrochemical approach was more beneficial than simple electrochemistry in most cases, probably because both single-electron oxidation and reduction are made possible at the same surface [45]. This is especially true for the radical cation Diels–Alder reaction, since non-substituted β-methylstyrene, which was previously reported as an unsuccessful dienophile, was found to participate under TiO2 photoelectrochemical conditions (Scheme 1) [46,47]. We questioned whether the scope of the radical cation Diels–Alder reaction could be further expanded, with particular interest on the installation of a second substituent at the β-position. Described herein is our unexpected finding that various spiro ring systems can be constructed by a radical cation Diels–Alder reaction of arylidene cycloalkanes.
![[1860-5397-18-112-i1]](/bjoc/content/inline/1860-5397-18-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Radical cation Diels–Alder reaction of β-methylstyrene.
Scheme 1: Radical cation Diels–Alder reaction of β-methylstyrene.
Results and Discussion
The present work began with the reaction of β-methylanethole (1) with 2,3-dimethyl-1,3-butadiene (2) under TiO2 photoelectrochemical and electrochemical conditions (Scheme 2). The initial attempts using both conditions provided us two small indications that the reaction was not totally inaccessible. The simple electrochemical approach gave a better result than TiO2 photoelectrochemistry. Furthermore, we confirmed that the additional methyl group at the β-position had a significant impact on the reaction. In general, tertiary radicals (or cations) are more stable than secondary ones and therefore, the additional methyl group seems to have a strong steric effect (Figure 3). If so, tying up the two methyl groups as a cyclopropane ring may decrease the steric hindrance at the β-position and improve the reaction. Unfortunately, the arylidene cyclopropane 4 was found to be totally unreactive under both conditions (Scheme 3). However, to our surprise, the arylidene cyclobutane 5 was found to be productive and the corresponding spiro ring compound 9 was obtained in good yield. The arylidene cyclopentane 6 and cyclohexane 7 were found to be less effective for the reaction and in particular, the former (6) was almost totally unsuccessful. Although the mechanism remains unclear, Knowles and Romanov-Michailidis recently reported that a similar trend is observed in photosensitized [2 + 2] cycloadditions [48]. Since their report was not a [4 + 2] but a [2 + 2] reaction and they proposed an energy transfer mechanism as opposed to an electron transfer pathway, it cannot be directly compared to our results. Even so, it would be fair to say that there is some correlation between these arylidene cycloalkane cycloadditions.
![[1860-5397-18-112-i2]](/bjoc/content/inline/1860-5397-18-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical cation Diels–Alder reaction of β-methylanethole (1). Recovered starting material is reported in parentheses.
Scheme 2: Radical cation Diels–Alder reaction of β-methylanethole (1). Recovered starting material is reporte...
![[1860-5397-18-112-3]](/bjoc/content/figures/1860-5397-18-112-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Formal expression of radical cations.
Figure 3: Formal expression of radical cations.
![[1860-5397-18-112-i3]](/bjoc/content/inline/1860-5397-18-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Radical cation Diels–Alder reactions of the arylidene cycloalkanes (4–7). Recovered starting material is reported in parentheses.
Scheme 3: Radical cation Diels–Alder reactions of the arylidene cycloalkanes (4–7). Recovered starting materi...
Control studies are summarized in Table 1. LiClO4, TiO2, and light were crucial for the reaction (Table 1, entries 1–4) and the equivalents of the diene 2 was also key (entries 5 and 6 in Table 1). The reaction was sensitive toward atmosphere; both oxygen and argon had a negative impact (Table 1, entries 7 and 8). In the electrochemical approach, potentiostatic conditions gave better results than galvanostatic conditions and more importantly, it was found that the reaction was completed within 0.5 F/mol (Table 1, entries 9–11). This result clearly suggests that a chain pathway is involved in the reaction.
Table 1: Control studies for the radical cation Diels–Alder reaction of the arylidene cyclobutane 5.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-112-i5.svg?max-width=637&scale=1.0)
|
||
| entry | deviation from the standard conditions | yield (%)a |
| 1 | 61 (0) | |
| 2 | no LiClO4 | 0 (40) |
| 3 | no light | 0 (83) |
| 4 | no TiO2 | 15 (32) |
| 5 | 2 equiv of diene | 22 (0) |
| 6 | 10 equiv of diene | 58 (0) |
| 7 | under O2 | 19 (16) |
| 8 | under Ar | 21 (0) |
| 9 | 1.3 V vs. Ag/AgCl, 0.1 F/mol | 22 (52) |
| 10 | 1.3 V vs. Ag/AgCl, 0.5 F/mol | 66 (0) |
| 11 | 1.0 mA, 0.5 F/mol | 46 (trace) |
arecovered starting material is reported in parentheses.
The scope of the reaction was studied using dimethyl and non-substituted aryl rings in combination with several cycloalkanes (Scheme 4). The TiO2 photoelectrochemical approach was more beneficial than simple electrochemistry in many cases for these dienophiles, which accords well with our previous reports. The ring size effect of cycloalkanes was also clearly observed and cyclobutane was much more effective than the others. A similar trend was observed using some heterocycles, which also accorded well with the previous report by Knowles and Romanov-Michailidis.
![[1860-5397-18-112-i4]](/bjoc/content/inline/1860-5397-18-112-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scope of the radical cation Diels–Alder reaction of arylidene cycloalkanes (recovered starting material is reported in parentheses).
Scheme 4: Scope of the radical cation Diels–Alder reaction of arylidene cycloalkanes (recovered starting mate...
Conclusion
In conclusion, we have demonstrated that radical cation Diels–Alder reactions of arylidene cycloalkanes are enabled under TiO2 photoelectrochemical and electrochemical conditions to construct various spiro ring systems. Although further detailed experimental and/or theoretical studies are required to elucidate the complete mechanistic picture, arylidene cyclobutanes were found to be much more effective than others. A similar ring size effect was observed by Knowles and Romanov-Michailidis in photosensitized [2 + 2] cycloadditions of benzylidene cycloalkanes and therefore, the results described herein may support a detailed mechanistic understanding. Further experimental and theoretical studies of radical cation cycloadditions of arylidene cycloalkanes are under investigation in our laboratory.
Experimental
Photoelectrochemical: The appropriate arylidene cycloalkane (0.20 mmol), 2,3-dimethyl-1,3-butadiene (2, 113 μL, 1.0 mmol), and TiO2 (100 mg) were added to a solution of LiClO4/CH3NO2 (1.0 M, 4.0 mL) while stirring at room temperature. The resulting reaction mixture was stirred at room temperature in front of a 15 W UV lamp (365 nm). Then, the solution was diluted with water and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Yields were determined by 1H NMR analysis using dibromomethane as an internal standard. Silica gel column chromatography (hexane/ethyl acetate) gave the corresponding spiro ring compound.
Electrochemical: The appropriate arylidene cycloalkane (0.20 mmol) and 2,3-dimethyl-1,3-butadiene (2, 113 μL, 1.0 mmol) were added to a solution of LiClO4/CH3NO2 (1.0 M, 4.0 mL) while stirring at room temperature. The resulting reaction mixture was electrolyzed at 1.3–1.5 V vs Ag/AgCl using carbon felt electrodes (10 mm × 10 mm) in an undivided cell with stirring. Then, the solution was diluted with water and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Yields were determined by 1H NMR analysis using dibromomethane as an internal standard. Silica gel column chromatography (hexane/ethyl acetate) gave the corresponding spiro ring compound.
Supporting Information
| Supporting Information File 1: General remarks, photocatalyst analyzation data, synthesis procedure, additional control studies, electrochemical measurements, and characterization data, including copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 8.1 MB | Download |
References
-
Bellville, D. J.; Wirth, D. W.; Bauld, N. L. J. Am. Chem. Soc. 1981, 103, 718–720. doi:10.1021/ja00393a061
Return to citation in text: [1] -
Bauld, N. L.; Bellville, D. J.; Harirchian, B.; Lorenz, K. T.; Pabon, R. A., Jr.; Reynolds, D. W.; Wirth, D. D.; Chiou, H. S.; Marsh, B. K. Acc. Chem. Res. 1987, 20, 371–378. doi:10.1021/ar00142a003
Return to citation in text: [1] -
Bauld, N. L. Tetrahedron 1989, 45, 5307–5363. doi:10.1016/s0040-4020(01)89486-2
Return to citation in text: [1] -
Mlcoch, J.; Steckhan, E. Tetrahedron Lett. 1987, 28, 1081–1084. doi:10.1016/s0040-4039(00)95916-1
Return to citation in text: [1] -
Gieseler, A.; Steckhan, E.; Wiest, O.; Knoch, F. J. Org. Chem. 1991, 56, 1405–1411. doi:10.1021/jo00004a013
Return to citation in text: [1] -
Haberl, U.; Wiest, O.; Steckhan, E. J. Am. Chem. Soc. 1999, 121, 6730–6736. doi:10.1021/ja983993y
Return to citation in text: [1] -
Valley, N. A.; Wiest, O. J. Org. Chem. 2007, 72, 559–566. doi:10.1021/jo0620361
Return to citation in text: [1] -
Pérez-Ruiz, R.; Domingo, L. R.; Jiménez, M. C.; Miranda, M. A. Org. Lett. 2011, 13, 5116–5119. doi:10.1021/ol201984s
Return to citation in text: [1] -
Lim, H. N.; Parker, K. A. J. Org. Chem. 2014, 79, 919–926. doi:10.1021/jo402082y
Return to citation in text: [1] -
Moore, J. C.; Davies, E. S.; Walsh, D. A.; Sharma, P.; Moses, J. E. Chem. Commun. 2014, 50, 12523–12525. doi:10.1039/c4cc05906a
Return to citation in text: [1] -
Tan, J. S. J.; Hirvonen, V.; Paton, R. S. Org. Lett. 2018, 20, 2821–2825. doi:10.1021/acs.orglett.8b00737
Return to citation in text: [1] -
Ischay, M. A.; Yoon, T. P. Eur. J. Org. Chem. 2012, 3359–3372. doi:10.1002/ejoc.201101071
Return to citation in text: [1] -
Studer, A.; Curran, D. P. Nat. Chem. 2014, 6, 765–773. doi:10.1038/nchem.2031
Return to citation in text: [1] -
Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem. 2014, 12, 6059–6071. doi:10.1039/c4ob00843j
Return to citation in text: [1] -
Luca, O. R.; Gustafson, J. L.; Maddox, S. M.; Fenwick, A. Q.; Smith, D. C. Org. Chem. Front. 2015, 2, 823–848. doi:10.1039/c5qo00075k
Return to citation in text: [1] -
Qiu, G.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 1011–1027. doi:10.1039/c6qo00103c
Return to citation in text: [1] -
Francke, R.; Little, R. D. ChemElectroChem 2019, 6, 4373–4382. doi:10.1002/celc.201900432
Return to citation in text: [1] -
Costentin, C.; Savéant, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 11147–11152. doi:10.1073/pnas.1904439116
Return to citation in text: [1] -
Reynolds, D. W.; Bauld, N. L. Tetrahedron 1986, 42, 6189–6194. doi:10.1016/s0040-4020(01)88079-0
Return to citation in text: [1] -
Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579
Return to citation in text: [1] -
Stevenson, S. M.; Shores, M. P.; Ferreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 6506–6510. doi:10.1002/anie.201501220
Return to citation in text: [1] -
Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723
Return to citation in text: [1] -
Alpers, D.; Gallhof, M.; Stark, C. B. W.; Brasholz, M. Chem. Commun. 2016, 52, 1025–1028. doi:10.1039/c5cc08994h
Return to citation in text: [1] -
Zhao, Y.; Antonietti, M. Angew. Chem., Int. Ed. 2017, 56, 9336–9340. doi:10.1002/anie.201703438
Return to citation in text: [1] -
Stevenson, S. M.; Higgins, R. F.; Shores, M. P.; Ferreira, E. M. Chem. Sci. 2017, 8, 654–660. doi:10.1039/c6sc03303b
Return to citation in text: [1] -
Yang, Y.; Liu, Q.; Zhang, L.; Yu, H.; Dang, Z. Organometallics 2017, 36, 687–698. doi:10.1021/acs.organomet.6b00886
Return to citation in text: [1] -
Shin, J. H.; Seong, E. Y.; Mun, H. J.; Jang, Y. J.; Kang, E. J. Org. Lett. 2018, 20, 5872–5876. doi:10.1021/acs.orglett.8b02541
Return to citation in text: [1] -
Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058
Return to citation in text: [1] -
Farney, E. P.; Chapman, S. J.; Swords, W. B.; Torelli, M. D.; Hamers, R. J.; Yoon, T. P. J. Am. Chem. Soc. 2019, 141, 6385–6391. doi:10.1021/jacs.9b01885
Return to citation in text: [1] -
Huber, N.; Li, R.; Ferguson, C. T. J.; Gehrig, D. W.; Ramanan, C.; Blom, P. W. M.; Landfester, K.; Zhang, K. A. I. Catal. Sci. Technol. 2020, 10, 2092–2099. doi:10.1039/d0cy00016g
Return to citation in text: [1] -
Tanaka, K.; Kishimoto, M.; Tanaka, Y.; Kamiyama, Y.; Asada, Y.; Sukekawa, M.; Ohtsuka, N.; Suzuki, T.; Momiyama, N.; Honda, K.; Hoshino, Y. J. Org. Chem. 2022, 87, 3319–3328. doi:10.1021/acs.joc.1c02972
Return to citation in text: [1] -
Tang, M.; Cameron, L.; Poland, E. M.; Yu, L.-J.; Moggach, S. A.; Fuller, R. O.; Huang, H.; Sun, J.; Thickett, S. C.; Massi, M.; Coote, M. L.; Ho, C. C.; Bissember, A. C. Inorg. Chem. 2022, 61, 1888–1898. doi:10.1021/acs.inorgchem.1c02964
Return to citation in text: [1] -
Yu, Y.; Fu, Y.; Zhong, F. Green Chem. 2018, 20, 1743–1747. doi:10.1039/c8gc00299a
Return to citation in text: [1] -
Horibe, T.; Ohmura, S.; Ishihara, K. J. Am. Chem. Soc. 2019, 141, 1877–1881. doi:10.1021/jacs.8b12827
Return to citation in text: [1] -
Horibe, T.; Ishihara, K. Chem. Lett. 2020, 49, 107–113. doi:10.1246/cl.190790
Return to citation in text: [1] -
Okada, Y.; Chiba, K. Chem. Rev. 2018, 118, 4592–4630. doi:10.1021/acs.chemrev.7b00400
Return to citation in text: [1] -
Okada, Y. Electrochemistry 2020, 88, 497–506. doi:10.5796/electrochemistry.20-00088
Return to citation in text: [1] -
Shida, N.; Imada, Y.; Okada, Y.; Chiba, K. Eur. J. Org. Chem. 2020, 570–574. doi:10.1002/ejoc.201901576
Return to citation in text: [1] -
Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/c7cc00664k
Return to citation in text: [1] -
Okada, Y.; Yamaguchi, Y.; Ozaki, A.; Chiba, K. Chem. Sci. 2016, 7, 6387–6393. doi:10.1039/c6sc02117d
Return to citation in text: [1] -
Okada, Y.; Maeta, N.; Nakayama, K.; Kamiya, H. J. Org. Chem. 2018, 83, 4948–4962. doi:10.1021/acs.joc.8b00738
Return to citation in text: [1] -
Okada, Y. J. Org. Chem. 2019, 84, 1882–1886. doi:10.1021/acs.joc.8b02861
Return to citation in text: [1] -
Okada, Y.; Yamaguchi, Y.; Chiba, K. ChemElectroChem 2019, 6, 4165–4168. doi:10.1002/celc.201900184
Return to citation in text: [1] -
Maeta, N.; Kamiya, H.; Okada, Y. Org. Lett. 2019, 21, 8519–8522. doi:10.1021/acs.orglett.9b02808
Return to citation in text: [1] -
Okada, Y. Chem. Rec. 2021, 21, 2223–2238. doi:10.1002/tcr.202100029
Return to citation in text: [1] -
Nakayama, K.; Maeta, N.; Horiguchi, G.; Kamiya, H.; Okada, Y. Org. Lett. 2019, 21, 2246–2250. doi:10.1021/acs.orglett.9b00526
Return to citation in text: [1] -
Nakayama, K.; Kamiya, H.; Okada, Y. J. Electrochem. Soc. 2020, 167, 155518. doi:10.1149/1945-7111/abb97f
Return to citation in text: [1] -
Murray, P. R. D.; Bussink, W. M. M.; Davies, G. H. M.; van der Mei, F. W.; Antropow, A. H.; Edwards, J. T.; D’Agostino, L. A.; Ellis, J. M.; Hamann, L. G.; Romanov-Michailidis, F.; Knowles, R. R. J. Am. Chem. Soc. 2021, 143, 4055–4063. doi:10.1021/jacs.1c01173
Return to citation in text: [1]
| 1. | Bellville, D. J.; Wirth, D. W.; Bauld, N. L. J. Am. Chem. Soc. 1981, 103, 718–720. doi:10.1021/ja00393a061 |
| 2. | Bauld, N. L.; Bellville, D. J.; Harirchian, B.; Lorenz, K. T.; Pabon, R. A., Jr.; Reynolds, D. W.; Wirth, D. D.; Chiou, H. S.; Marsh, B. K. Acc. Chem. Res. 1987, 20, 371–378. doi:10.1021/ar00142a003 |
| 3. | Bauld, N. L. Tetrahedron 1989, 45, 5307–5363. doi:10.1016/s0040-4020(01)89486-2 |
| 4. | Mlcoch, J.; Steckhan, E. Tetrahedron Lett. 1987, 28, 1081–1084. doi:10.1016/s0040-4039(00)95916-1 |
| 5. | Gieseler, A.; Steckhan, E.; Wiest, O.; Knoch, F. J. Org. Chem. 1991, 56, 1405–1411. doi:10.1021/jo00004a013 |
| 6. | Haberl, U.; Wiest, O.; Steckhan, E. J. Am. Chem. Soc. 1999, 121, 6730–6736. doi:10.1021/ja983993y |
| 7. | Valley, N. A.; Wiest, O. J. Org. Chem. 2007, 72, 559–566. doi:10.1021/jo0620361 |
| 8. | Pérez-Ruiz, R.; Domingo, L. R.; Jiménez, M. C.; Miranda, M. A. Org. Lett. 2011, 13, 5116–5119. doi:10.1021/ol201984s |
| 9. | Lim, H. N.; Parker, K. A. J. Org. Chem. 2014, 79, 919–926. doi:10.1021/jo402082y |
| 10. | Moore, J. C.; Davies, E. S.; Walsh, D. A.; Sharma, P.; Moses, J. E. Chem. Commun. 2014, 50, 12523–12525. doi:10.1039/c4cc05906a |
| 11. | Tan, J. S. J.; Hirvonen, V.; Paton, R. S. Org. Lett. 2018, 20, 2821–2825. doi:10.1021/acs.orglett.8b00737 |
| 36. | Okada, Y.; Chiba, K. Chem. Rev. 2018, 118, 4592–4630. doi:10.1021/acs.chemrev.7b00400 |
| 37. | Okada, Y. Electrochemistry 2020, 88, 497–506. doi:10.5796/electrochemistry.20-00088 |
| 38. | Shida, N.; Imada, Y.; Okada, Y.; Chiba, K. Eur. J. Org. Chem. 2020, 570–574. doi:10.1002/ejoc.201901576 |
| 39. | Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/c7cc00664k |
| 40. | Okada, Y.; Yamaguchi, Y.; Ozaki, A.; Chiba, K. Chem. Sci. 2016, 7, 6387–6393. doi:10.1039/c6sc02117d |
| 41. | Okada, Y.; Maeta, N.; Nakayama, K.; Kamiya, H. J. Org. Chem. 2018, 83, 4948–4962. doi:10.1021/acs.joc.8b00738 |
| 42. | Okada, Y. J. Org. Chem. 2019, 84, 1882–1886. doi:10.1021/acs.joc.8b02861 |
| 43. | Okada, Y.; Yamaguchi, Y.; Chiba, K. ChemElectroChem 2019, 6, 4165–4168. doi:10.1002/celc.201900184 |
| 44. | Maeta, N.; Kamiya, H.; Okada, Y. Org. Lett. 2019, 21, 8519–8522. doi:10.1021/acs.orglett.9b02808 |
| 33. | Yu, Y.; Fu, Y.; Zhong, F. Green Chem. 2018, 20, 1743–1747. doi:10.1039/c8gc00299a |
| 34. | Horibe, T.; Ohmura, S.; Ishihara, K. J. Am. Chem. Soc. 2019, 141, 1877–1881. doi:10.1021/jacs.8b12827 |
| 35. | Horibe, T.; Ishihara, K. Chem. Lett. 2020, 49, 107–113. doi:10.1246/cl.190790 |
| 19. | Reynolds, D. W.; Bauld, N. L. Tetrahedron 1986, 42, 6189–6194. doi:10.1016/s0040-4020(01)88079-0 |
| 20. | Lin, S.; Ischay, M. A.; Fry, C. G.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. doi:10.1021/ja2093579 |
| 21. | Stevenson, S. M.; Shores, M. P.; Ferreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 6506–6510. doi:10.1002/anie.201501220 |
| 22. | Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723 |
| 23. | Alpers, D.; Gallhof, M.; Stark, C. B. W.; Brasholz, M. Chem. Commun. 2016, 52, 1025–1028. doi:10.1039/c5cc08994h |
| 24. | Zhao, Y.; Antonietti, M. Angew. Chem., Int. Ed. 2017, 56, 9336–9340. doi:10.1002/anie.201703438 |
| 25. | Stevenson, S. M.; Higgins, R. F.; Shores, M. P.; Ferreira, E. M. Chem. Sci. 2017, 8, 654–660. doi:10.1039/c6sc03303b |
| 26. | Yang, Y.; Liu, Q.; Zhang, L.; Yu, H.; Dang, Z. Organometallics 2017, 36, 687–698. doi:10.1021/acs.organomet.6b00886 |
| 27. | Shin, J. H.; Seong, E. Y.; Mun, H. J.; Jang, Y. J.; Kang, E. J. Org. Lett. 2018, 20, 5872–5876. doi:10.1021/acs.orglett.8b02541 |
| 28. | Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058 |
| 29. | Farney, E. P.; Chapman, S. J.; Swords, W. B.; Torelli, M. D.; Hamers, R. J.; Yoon, T. P. J. Am. Chem. Soc. 2019, 141, 6385–6391. doi:10.1021/jacs.9b01885 |
| 30. | Huber, N.; Li, R.; Ferguson, C. T. J.; Gehrig, D. W.; Ramanan, C.; Blom, P. W. M.; Landfester, K.; Zhang, K. A. I. Catal. Sci. Technol. 2020, 10, 2092–2099. doi:10.1039/d0cy00016g |
| 31. | Tanaka, K.; Kishimoto, M.; Tanaka, Y.; Kamiyama, Y.; Asada, Y.; Sukekawa, M.; Ohtsuka, N.; Suzuki, T.; Momiyama, N.; Honda, K.; Hoshino, Y. J. Org. Chem. 2022, 87, 3319–3328. doi:10.1021/acs.joc.1c02972 |
| 32. | Tang, M.; Cameron, L.; Poland, E. M.; Yu, L.-J.; Moggach, S. A.; Fuller, R. O.; Huang, H.; Sun, J.; Thickett, S. C.; Massi, M.; Coote, M. L.; Ho, C. C.; Bissember, A. C. Inorg. Chem. 2022, 61, 1888–1898. doi:10.1021/acs.inorgchem.1c02964 |
| 12. | Ischay, M. A.; Yoon, T. P. Eur. J. Org. Chem. 2012, 3359–3372. doi:10.1002/ejoc.201101071 |
| 13. | Studer, A.; Curran, D. P. Nat. Chem. 2014, 6, 765–773. doi:10.1038/nchem.2031 |
| 14. | Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem. 2014, 12, 6059–6071. doi:10.1039/c4ob00843j |
| 15. | Luca, O. R.; Gustafson, J. L.; Maddox, S. M.; Fenwick, A. Q.; Smith, D. C. Org. Chem. Front. 2015, 2, 823–848. doi:10.1039/c5qo00075k |
| 16. | Qiu, G.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 1011–1027. doi:10.1039/c6qo00103c |
| 17. | Francke, R.; Little, R. D. ChemElectroChem 2019, 6, 4373–4382. doi:10.1002/celc.201900432 |
| 18. | Costentin, C.; Savéant, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 11147–11152. doi:10.1073/pnas.1904439116 |
| 48. | Murray, P. R. D.; Bussink, W. M. M.; Davies, G. H. M.; van der Mei, F. W.; Antropow, A. H.; Edwards, J. T.; D’Agostino, L. A.; Ellis, J. M.; Hamann, L. G.; Romanov-Michailidis, F.; Knowles, R. R. J. Am. Chem. Soc. 2021, 143, 4055–4063. doi:10.1021/jacs.1c01173 |
| 46. | Nakayama, K.; Maeta, N.; Horiguchi, G.; Kamiya, H.; Okada, Y. Org. Lett. 2019, 21, 2246–2250. doi:10.1021/acs.orglett.9b00526 |
| 47. | Nakayama, K.; Kamiya, H.; Okada, Y. J. Electrochem. Soc. 2020, 167, 155518. doi:10.1149/1945-7111/abb97f |
© 2022 Nakayama et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.